1.Предмет фармацевтической химии.Области исследования и связь с другими науками.
Фармацевтическая химия -- наука, которая, базируясь на общих законах химических наук, исследует способы получения, строение, физические и химические свойства лекарственных веществ, взаимосвязь между их химической структурой и действием на организм, методы контроля качества и изменения, происходящие при хранении.
Основными методами исследования лекарственных веществ в фармацевтической химии являются анализ и синтез -- диалектически тесно связанные между собой процессы, взаимно дополняющие друг друга. Фармацевтическая химия является важным разделом химической науки и тесно связана с ее отдельными дисциплинами. Используя достижения базовых химических дисциплин, фармацевтическая химия решает задачу целенаправленного поиска новых лекарственных средств. Например, современные компьютерные методы позволяют прогнозировать фармакологическое действие (терапевтический эффект) лекарственного средства.Фармакогнозия -- наука, изучающая лекарственное растительное сырье и возможности создания из него новых лекарственных веществ. Тесно взаимосвязана фармацевтическая химия с фармацевтической технологией, изучающей методы приготовления лекарственных средств. Они являются объектами для разработки способов фармацевтического анализа. Токсикологическая химия базируется на применении целого ряда тех же методов исследования, что и фармацевтическая химия. В изучении проблем хранения лекарственных средств, а также организации контрольно-аналитической службы тесно связаны с фармацевтической химией организация и экономика фармации. В области исследования взаимосвязи между структурой молекул лекарственных веществ и их действием на организм фармацевтическая химия близко примыкает к фармакологии.Вместе с тем фармацевтическая химия занимает промежуточное положение между комплексом медико-биологических и химических наук. Объектом применения лекарственных средств является организм больного человека.Тесная взаимосвязь со всеми перечисленными дисциплинами обеспечивает решение современных проблем фармацевтической химии.
2.Основные этапы истории фармхимии. Современные проблемы и перспективы развития фармхиии.
Период ятрохимии (XVI -- XVII вв.). В эпоху возрождения на смену алхимии пришла ятрохимия (лечебная химия). Ее основатель Парацельс (1493 -- 1541) считал, что "не добыванию золота, а защите здоровья должна служить химия".Парацельс провел исследование действия на организм многих веществ минерального и растительного происхождения. Он усовершенствовал ряд приборов и аппаратов для выполнения анализа. Вот почему Парацельса по праву считают одним из основоположников фармацевтического анализа, а ятрохимию -- периодом зарождения фармацевтической химии.
Аптеки в XVI -- XVII вв. были своеобразными центрами по изучению химических веществ. В них получали и исследовали вещества минерального, растительного и животного происхождения.
Период зарождения первых химических теорий (ХVII -- XIX вв.). Для развития промышленного производства в этот период необходимо было расширить рамки химических исследований за пределы иатрохимии. Это привело к созданию первых химических производств и к формированию химической науки.
Вторая половина XVII в. --период зарождения первой химической теории -- теории флогистона. С ее помощью пытались доказать, что процессы горения и окисления сопровождаются выделением особого вещества -- "флогистона". Теорию флогистона создали И.Бехер (1635-1682) и Г.Шталь (1660-1734).
Многое сделал для развития фармацевтического анализа аптекарь Мор (1806 -- 1879). Он впервые применил бюретки, пипетки, аптечные весы, которые носят его имя.
Таким образом, фармацевтическая химия, зародившаяся в период ятрохимии в XVI в., получила свое дальнейшее развитие в XVII -- XVIII вв.
Современные проблемы фармацевтической химии
Основными проблемами фармацевтической химии являются:
- создание и исследование новых лекарственных средств;
- разработка способов фармацевтического и биофармацевтического анализа.
Создание и исследование новых ЛС. Несмотря на огромный арсенал имеющихся ЛС, проблема изыскания новых высокоэффективных ЛВ остается актуальной.
Роль ЛС непрерывно растет в современной медицине. Это вызвано целым рядом причин, главными из которых являются:
- ряд тяжелых заболеваний еще не излечивается ЛС;
- длительное применение ряда ЛС формирует толерантные патологии, для борьбы с которыми необходимы новые ЛС с иным механизмом действия;
- процессы эволюции микроорганизмов приводят к возникновению новых заболеваний, для лечения которых нужны эффективные ЛС;
- некоторые из применяемых ЛВ вызывают побочные эффекты, в связи с чем необходимо создавать более безопасные ЛС.
Таким образом, современная номенклатура ЛC в различных фармакотерапевтических группах требует дальнейшего расширения. Создаваемые новые ЛC только в том случае являются перспективными, если по своей эффективности и безопасности они превосходят существующие, а по качеству соответствуют мировым требованиям. В решении этой проблемы важная роль принадлежит специалистам в области фармацевтической химии, которая отражает общественно-медицинскую значимость этой науки.
3.Терминология:ЛС,субстанция для фармацевтического использования,ЛФ,гомеопатическиеЛС,оригинальноеЛС,генерическоеЛС,иммуннобиологич
еское,радиофармацевтическое.
Биодоступность- полнота и скорость всасывания ЛВ, которые характеризуются его количеством,поступившим в организм, после применения ЛП.
Биоэквивалентность -равенство биодоступности в допустимых пределах одних и тех же ЛП, приготовленных разными производителями.
Валидация-оценка и дкументальное подтверждение соответствия производственного процесса и качества продукции утвержжденным требованиям.
Вспомогательное вещество-относительно индефферентное в химич-о и биолоч-о отношении в-во, разрешенное к медицинскому применению в целях получения ЛФ, придания или сохранения определенывх свойств ЛП.
Качество ЛП-совокупность свойств, которые придают ЛП способность соответствовать своему назначению и отвечают требованиям НД.
Лекарственная форма - состояние, придаваемое ЛС или лекарственному растительному сырью, удобное для применения, обеспечивающее необходимый лечебный эффект.
Лекарственное (фармацевтическое) сырье - ЛС, лекарственное растительное сырье, вспомогательные вещества, разрешенные к медицинскому применению для производства лекарственных препаратов или другой фармацевтической продукции или полуфабрикатов. Фактически понятие «сырье» включает все исходные материалы, поступающие в производство для переработки с целью получения готового продукта или полуфабриката.
Фармацевтические субстанции - лекарственные средства в виде действующих веществ биологического, биотехнологического, минерального или химического происхождения, обладающие фармакологической активностью, предназначенные для производства, изготовления лекарственных препаратов и определяющие их эффективность.
Лекарственные средства- вещества, применяемые для профилактики, диагностики и лечения болезни, предотвращения беременности, полученные из крови, плазмы крови, а также органов, тканей человека или животного, растений, микроорганизмов, минералов методами синтеза или с применением биологических технологий.
Лекарственные препараты - дозированные ЛС в определенной лекарственной форме, готовые к применению.
Оригинальные лекарственные средства - ЛС, поступившие в обращение с зарегистрированными собственными названиями.
Воспроизведенные ЛС (дженерики) - ЛС, поступившие в обращение после истечения срока действия исключительных патентных прав на оригинальные ЛС.
Радиоактивное средство - ЛС, применяемое в медицинской практике в связи с его способностью к ионизирующему излучению.
Сильнодействующее средство- ЛС с высокой биологической активностью, прописывание, отпуск, хранение и учет которого производятся по особым правилам, установленным Минздравом России.
Наркотическое средство-ядовитое или сильнодействующее ЛС,требующего ограниченного применения и отнесенное к НС в соответствии с законодательством.
4.Правила выбора названий ЛС.МНН фармацевтических субстанций.
Лекарственные средства, как правило, имеют по несколько наименований (названий). Число синонимов синтетического лекарственного вещества достигает нескольких десятков и даже сотен. Химическое название отражает структуру JIB и присваивается в соответствии с правилами международной химической терминологии.
Однозначное название, как правило, имеют алкалоиды (пилокарпин, морфин, атропин). Они даются исходя кз наименований производящих растений. Аналогично происхождение названий других БАВ растительного и животного происхождения, в т.ч. гликозидов, ферментов, гормонов (инсулин, кортизон, тестостерон). Наименования JIC из числа антибиотиков происходят от их продуцентов (пенициллин, цефалоспорин). Целый ряд названий синтетических ЛС формируется из слогов их полного химического названия (парацетамол, промедол, хлорпромазин, нифедипин и др.). Нередко название присваивается на основе терапевтического действия (панадол, спазмолитин, апрессин, анальгин и др.). Иногда сочетаются в названии элементы химического строения и терапевтического действия. Некоторые производители включают в наименование часть названия фирмы.
Одним из важнейших направлений стандартизации JIC, которые регистрируются в Российской Федерации, является правильность присвоения им названий.
Комиссия по международным названиям ВОЗ с целью упорядочения и унификации названийJIC во всех странах мира разработала международную классификацию, в основу которой заложена определенная система формирования терминологии JIB. Принцип этой системы INN — МНН (InternationalNonproprietaryNames — международные непатентованные наименования) заключается в том, что в названии JIB ориентировочно дается его групповая принадлежность.
Многим отечественным JIB также присвоено МНН. Однако целый ряд из них имеют традиционную для России латинскую терминологию (Resorcinum, Mentholum), которая сохранилась в НД. Поэтому при изучении фармацевтической химии будет использована в основном номенклатура МНН, а при ее отсутствии — сохранившиеся латинские названия. В качестве основного синонима будут также приводиться торговые названия, под которыми JIC зарегистрировано или производится в Российской Федерации.
5.Принципы классификации ЛС, используемые в фармацевтической химии.
Известно несколько типов калссификации ЛС.
1)Химическая классификация разделяет ЛС на 2 большие группы:ЛС неорганической природы:в соответствии с положениями в периодической системе элементов,к ним относятся препараты s-,p-,d-элементов. ЛС органической природы :к ним относятся органическрие соединения, имеющие ту или иную структуру и содержащие те или иные функциональные группы. Напр :алифатические соединения среди прочих включают Лсальдегидов, эфиров, аминокислот;к ароматическим относятся фенолы,ароматическиекислоты.ЛС органической природы могут быть разделны на подгруппы в зависимости от способа получения :природные, синтетические, полусинтетические.
Фармаколлгическая классфикация отражает действие ЛП на ту или иную систему организма(сердечно-сосудистая , нервная).Внутри групп фармакологической классификации препаратов может рассматриваться фармакотерапевтическая классификация- ЛП группируются в зависимости от применения для лечения определенного заболевания (противомикробные, противовирусные, снотворные и т.д).
6.Источники и спососбы получения ЛС.
Для получения неорганических ЛС используется минеральное сырье(природные источники). Например, для приготовления ЛС натрия хлорида используются природные растворы-воды озер и морей. Синтетические органические ЛС получают из продуктов переработки каменного угля, нефти, дерева и т.д.Выделенные при этом индивидуальные органические соединения являются реагентами в органическом синтезе ЛВ.Так осуществлен полный химический синтез антибиотика левомицетина- и алкалоида кофеина. Источником получении органических ЛВ является лекарственное растительное сырье .Из него получают алкалоиды, терпены, гликозиды, витамины, ЭМ. Растительное сырье используют также для получения галеновых препаратов. Гормональные препараты готовят из сырья животного происхождения. Для получения антибиотиков используют различные микроорганизмы. Известны полусинтетические антибиотики, которые являются синтетическими производными антибиотков, выделнных и з микроорганизмов.(пенициллины).Полусинтетический способ используется и для получения алкалоидов, витаминов, гормонов. В ХХ в. Было создано большое число новых синтетических ЛС-противоопухолевых,гипотензивных,сердечно-сосудистых,антибиотиков,сульфаниламидных препаратов.Номенклатура ЛС растет с каждым годом.
7.Титриметрические методы анализа в фармацевтическом анализе. Классификация методов.
Титриметрия- это метод количественного анализа, основанный на точном измерении объема раствора реактива известной концентрации, затраченного на химическую реакцию с определяемым веществом. Раствор реагента с точно известной концентрацией называется
стандартным раствором, или титрантом. Процесс постепенного приливания раствора титранта к раствору анализируемого вещества называют титрованием. По способу выполнения различают прямое, обратное титрование и титрование заместителя. При прямом титровании титрант непосредственно добавляют к титруемому веществу. В случае обратного титрования к определяемому веществу добавляют заведомый избыток титранта, реагирующего с исследуемым веществом в стехиометрическом количестве, проводят реакцию до конца, а затем избыток непрореагировавшего титранта оттитровывают другим стандартным раствором. Этот способ используют, если скорость прямой реакции мала или не удается подобрать подходящий индикатор. Если реакция нестехиометрична или протекает очень медленно, применяют способ заместительного титрования. К анализируемому раствору добавляют вспомогательный реагент, с которым определяемый компонент реагирует стехиометрически, а получающийся в эквивалентном количестве продукт реакции (заместитель) оттитровывают подходящим титрантом.
Методом кислотно-основного
титрования (нейтрализации) определяют количество кислот
(алкалиметрия) или оснований (ациди-метрия) в данном растворе, количество солей
слабых кислот и слабых оснований, а также веществ, которые реагируют с этими солями.
Применение неводных растворителей (спирты, ацетон и т. п.) позволило расширить
круг веществ, которые можно определять данным методом. Методы осаждения и
комплексообразования. Сюда относятся титриметрические определения,
основанные на осаждении того или иного иона в виде малорастворимого соединения
или связывания его в мало диссоциированный комплекс. Методы
окисления — восстановления (редоксиметрия). Эти методы основаны на
реакциях окисления и восстановления. Их называют обычно по применяемому
титрованному раствору реагента, например:
перманганатометрия, в которой используются реакции окисления перманганатом
калия KMnO4;
иодометрия, в которой используются реакции окисления иодом или восстановления
I-ионами;
бихроматометрия, в которой используются реакции окисления бихроматом калия
К2Сr2О7;
броматометрия, в которой используются реакции окисления броматом калия КВrO3.
К методам окисления — восстановления относятся также цериметрия
(окисление Се4+-ионами), ванадатометрия (окисление VO3-ионами), титанометрия
(восстановление Т13+-ионами). По способу титрования различают следующие методы.
8.Методы осадительного титрования в фармации.Аргентометрия:метод Мора ,Фольгарда,метод Фаянса.
Метод осадительного титрования основан на применении реакций осаждения, в результате которых образуются малорастворимые соединения. Аргентометрия – метод объемного анализа, основанный на применении стандартного раствора (титранта) ионов серебра.
Метод Мора-определение галогенид-ионов прямым титрованием раствором нитрата серебра в присутствии индикатора -раствора хромата калия .применяется для определения хлоридов и бромидов.Этот метод не позволяет определить I и NCS,т.к при титровании происходит соосаждение хромата калия с осадками AgI,AgNCS.
Метод Фаянса-определение галогенид-ионов прямым титрованием раствором нитрата серебра в присутствии адсорбционных индикаторов-флуоресцеина ,эозина. Метод позволяет определить хлориды, бромиды, йодиды, цианиды, тиоцианаты.
Метод Фольгарда–обратное титрование избытка катионов серебра раствором тиоционата аммония NH4SCNили калия KSCNв присутсвии индикатора –соли железа, обычно, железоаммонийных квасцов.NH4Fe(SO4)2*12H2O
9.Условный титр и его вычисление.
Условный титр-масса раствореного вещества в граммах в одном миллилитре раствора .(все формулы для нахождения массовой доли при прямом ,обратном и заместительном титровании). (ТЕТРАДЬ)

________________________________________________________________________________
_________________________________________________________________________________
_________________________________________________________________________________
_________________________________________________________________________________
_________________________________________________________________________________
_________________________________________________________________________________
_________________________________________________________________________________
_________________________________________________________________________________
_________________________________________________________________________________
______________________________________________________________________________
10.Абсорбционные спектроскопические методы: молекулярная абсорбционная спектрометрия в УФ- и видимой областях в фармации.
Молекулярную абсорбционную спектроскопию в УФ- и видимой областях спектра называют спектрофотометрией либо фотометрией. Молеклярно-абсорбцинная спектроскопия в УФ и видимой областях основана на избирательном поглощении веществами излучения..Спектрофотометр сосотоит из: дифракционной решетки, диодной линейки, лампы, кюветы линзы. Спектрофотометрические определения: Определение содержания веществ можно проводить непосредственно,а также с использованием специальных фотометрических реагентов. Определение без реагентов. В УФ области можно определять многие органические соединения , такие как ароматичемские углеводороды и т.д.В видимой области можно определять окрашенные органические и неорганические соединения ,как перманганат и хромат ионы.
Определение с помощью реагентов. Для проведение анализа проводят фотометрическую реакцию.
Применение спектроскопии в УФ и видимой областях в фармацевтическом анализе.
1)Оценка подлинности ЛВ.
Идентификация на основании электронного спектра. Спектр ЛВ сравнивается со спектром стандартного образца этого вещества .Например, УФ-спектр фуросемида 0,0005% раствора фуросемида в 0,01М растворе NaOH имеет два максимума поглощения-при 228 и 271 нм и один минимум –при 249нм.,тогда ка 0,005%раствор фуросемида в том же растворителе содержит один максимум при 333нм и минимум при 295нм.
Идентификации на основании коэффициента поглощения. Например ,коэффициент поглощения парацетамола в 0,1М растворе НС1 и при 240 нм равен 880л/моль см.
2)оценка чистоты ЛВ. Готовят раствор ЛВ определенной концентрации и измеряют величину оптической плотности при определенной длине волны. Например, оптическая плотность 16% раствора анальгина при 400нм не должна превышать 0,1. Обнаружение специфических примесей по появлению дополнительных полос поглощения в электронном спектре. Например, максимум поглощения адреналина находится при 278нм,а его специфической примеси –адреналона при 310нм.
3)Определение количественного содержания ЛВ. Спектрофотометрической константой является удельный показатель поглощения,который расчитывают по формуле:Е1%1см=А/С*L
11.Рефрактометрия в фармацевтическом анализе.
Рефрактометрия – метод анализа лекарственных средств, основанный на определении показателя преломления испытуемого вещества. Показателем преломления (индексом рефракции) называют отношение скорости света в вакууме к скорости света в испытуемом веществе (абсолютный показатель преломления). На практике определяют так называемый относительный показатель преломления (n), который является отношением скорости света в воздухе к скорости света в испытуемом веществе.
Показатель преломления зависит от температуры и длины волны света, при которой проводят определение. В растворах показатель преломления зависит также от концентрации вещества и природы растворителя. Рефрактометрию применяют для установления подлинности и чистоты вещества. Метод применяют также для определения концентрации вещества в растворе, которую находят по графику зависимости показателя преломления раствора от концентрации раствора. На графике выбирают интервал концентраций, в котором наблюдается линейная зависимость между показателем преломления и концентрацией. В этом интервале концентрацию испытуемого раствора (Х, %) вычисляют по формуле: X = (n – no)/F,
Где : n – показатель преломления испытуемого раствора; nо – показатель преломления растворителя при той же температуре; F – фактор, равный величине прироста показателя преломления при увеличении концентрации испытуемого раствора на 1 % (устанавливается экспериментально).
Для определения показателя преломления применяют рефрактометры. Определение проводят при температуре (20 ± 0,5) ºС и длине волны линии D спектра натрия (589,3 нм). Показатель преломления, определенный при таких условиях, обозначается индексом n20D. Современные приборы откалиброваны таким образом, что отсчеты, полученные по их шкалам, соответствуют показателям преломления для D линии спектра натрия. При проведении измерений следует соблюдать указания в отношении соответствующего источника света, приведенные в инструкции к прибору. Если используют белый свет, то рефрактометр снабжен компенсирующей системой. Цена деления термометра не должна превышать 0,5 ºС. Обычно измерения показателя преломления проводят на рефрактометрах Аббе, в основу которых положено явление полного внутреннего отражения при прохождении светом границы раздела двух сред с разными показателями преломления. Диапазон измеряемых показателей преломления при измерении в проходящем свете 1,3 – 1,7. Точность измерения показателя преломления должна быть не ниже ± (2 · 10-4). Могут быть использованы рефрактометры других типов с такой же или большей точностью. Рефрактометры юстируют по эталонным жидкостям, значения показателей преломления которых обозначены на этикетке, или по дистиллированной воде, для которой n20D = 1,3330 и n25D = 1,3325 (Δn/Δ = – 0,000085).
12.Поляриметрический метод в фармации.
Поляриметрия- метод основанный на способности вещества вращать плоскость поляризованного света. Это способность обусловлена наличием в молекулах ассиметрических атомов углерода . Степень отклонения плоскости поляризации от первоначального полодения выражается в угловых градусах. Эту величину называют углом вращения(а).Правовращающие вещества вращают плоскость поляризации по часовой стрелке (+),левовращающие(-).
Для растворов величина а зависит от природы растворителя, концентрации оптически активного вещества и длины рабочего слоя кюветы с раствором. Подлинность и чистоту ЛВ подтверждают по величине удельного вращения [a]20d=а*100/L*C,где а-измеренный угол вращения, в градусах,1-длина рабочего слоя кюветы, вдм, С-концентрация р-ра в-ва (г/100мл).
Количественно определяют (в%)содержание оптически активных в-в в растворе по формуле:
С=а*100/(а)20d*L. Величину а измеряют на поляриметрах с точностью до +-0,02.
13.Потенциометрическое определение рН.
Водородным показателем (рH) называется отрицательный десятичный
логарифм активности ионов водорода. рН =log аH +
рН – число, характеризующее концентрацию (активность) ионов водорода в
водных растворах. На практике рН определяют экспериментально. рН исследуемого раствора связан с рН стандартного раствора (рНS) следующим уравнением: рН=PHs-E-Es/k
где: Е– потенциал электрода в исследуемом растворе, в вольтах; ES – потенциал того же электрода в растворе с известным рН (рНS), вольтах; k – температурный коэффициент.
Потенциометрическое определение рН проводят путем измерения разности потенциалов между двумя электродами, погруженными в исследуемый раствор: один из электродов чувствителен к ионам водорода (обычно это стеклянный электрод), другой – электрод сравнения (например, насыщенный каломельный электрод).
Прибор. В качестве прибора для потенциометрического определения рН используют иономеры или рН-метры с чувствительностью не менее 0,05 единиц рН или 3 мВ. Калибровка приборов производится по стандартным буферным растворам.
 Методика. Все измерения проводят
при одной и той же температуре в интервале от 20оС до 25оС, если отсутствуют
иные указания в частной статье. При необходимости используют температурные
поправки в соответствии с инструкцией предприятия-производителя. Прибор
калибруют с помощью буферного раствора калия гидрофталата (первичный стандарт)
и одного из буферных растворов с другим значением рН (желательно из одного,
указанных в Табл.2.2.3.-2). Показания прибора для третьего буферного раствора с
промежуточным значением рН не могут отличаться больше чем на 0.05 единиц рН от
табличного значения рН, соответствующего этому раствору. Электроды погружают в
исследуемый раствор и измеряют рН при тех же условиях, что и для буферных
растворов. Подготовка прибора, электродной системы, а также градуировка прибора
проводится согласно инструкциям, прилагаемым к прибору. Если прибор
используется ежедневно, его градуировку проводят регулярно (не реже одного раза
в неделю). В противном случае градуировку прибора проводят перед каждым
измерением. Все исследуемые растворы и стандартные буферные растворы должны
быть приготовлены на воде, не содержащей диоксида углерода Р.,
должна иметь рН 5.8-7.0.
Методика. Все измерения проводят
при одной и той же температуре в интервале от 20оС до 25оС, если отсутствуют
иные указания в частной статье. При необходимости используют температурные
поправки в соответствии с инструкцией предприятия-производителя. Прибор
калибруют с помощью буферного раствора калия гидрофталата (первичный стандарт)
и одного из буферных растворов с другим значением рН (желательно из одного,
указанных в Табл.2.2.3.-2). Показания прибора для третьего буферного раствора с
промежуточным значением рН не могут отличаться больше чем на 0.05 единиц рН от
табличного значения рН, соответствующего этому раствору. Электроды погружают в
исследуемый раствор и измеряют рН при тех же условиях, что и для буферных
растворов. Подготовка прибора, электродной системы, а также градуировка прибора
проводится согласно инструкциям, прилагаемым к прибору. Если прибор
используется ежедневно, его градуировку проводят регулярно (не реже одного раза
в неделю). В противном случае градуировку прибора проводят перед каждым
измерением. Все исследуемые растворы и стандартные буферные растворы должны
быть приготовлены на воде, не содержащей диоксида углерода Р.,
должна иметь рН 5.8-7.0.

14.Газовая хроматография в фармации.
Газожидкостная хроматография основана распределении компонентов смеси м/у газовой жидкой или твердыми фазами. Распределение происходит в результате многократных актов сорбции и десорбции анализируемых в-в ,которые вводятся в поток газа-носителя, испаряются и в парообразном состоянии проходят ч/з колонку с сорбентом. Поэтому метод ГЖХ пименим для анализа летучих веществ или в-в ,которые могут быть переведены в газообразное состояние .Разделенные вещества элюируются из колонки потоком газа-носителя, регистрируются детектором и фиксируются на хроматограмме в виде пиков ,по которым можно идентифицировать или определять содержание каждого компонента смеси.
Газовый хроматограф включает в себя систему измерения и регулирования скорости потока газа-носителя, систему ввода пробы испытуемого в-ва, газохроматографическую колонку ,систему термостатирования и контроля температуры в различных узлах прибора и схему детектирования , регистрации и обработки информации ,полученной на приборе.
Подлинность ЛВ методом ГЖХ можно подтвердить либо с помощью свидетелей ,либо методом относительных удерживаний. В первом случае доказательством идентичности служит совпадение времени удерживания в-ва -свидетеля и одного из компонентов смеси ЛВ при хроматографировании каждого в отдельности в одинаковых условиях. Во втором случае в-во-свидетель добавляют к пробе ,затеи анализируют по рекомендуемой методике. Рассчитывают по формуле велоичину относительного удерживания, которая является постоянной для ЛВ в конкретных условиях. Количественный анализ выполняют в тех же условиях ,используя для расчетов такие параметры, как площадь или высота пиков ЛВ. Площадь пиков устанавливают на хроматограмме с помощью планиметра, интегратора, или умножением высоты пика на его полуширину.
15. Тонкослойная хроматография и ее применение в фармацевтическом анализе.
ТСХ отличается от хроматографии на бумаге тем , что процесс хроматографировпания происходит на носителе (сорбенте),нанесенном тонким слоем на инертную поверхность. Твердый сорбент может быть закрепленным или незакрепленным на этой поверхности. Сорбентом служить силикогель или оксид алюминия Для закрепления добавляют небольшие количества крахмал ил сульфата кальция. Используют также пластинки промышленного изготовления типа «Силуфол УФ-254», «Сорбил».
Преимуществом ТСХ является простота приемов и оборудования, более и высокая чувствительность ,чем у бумажной хроматографии ,устойчивость пластинок к температурным и химическим воздействиям .меньшая продолжительность выполнения испытания. Все это создает широкие возможности в использовании ТСХ для выполнения испытаний на подлинность ,на чистоту ,для количественного определения ЛВ в ЛФ. В фармацевтическом анализе широко применяют сочетание ТСХ с физико-химическими методами анализа. Такие комбинированные методы, как хромато-спектрофотометрия,хромато-флуориметрия,особенноэфффективны в анализе ЛРС и препаратов ,содержащих большое количество сопутствующих компонентов.
16. Высокоэффективная жидкостная хроматография (ВЭЖХ).
Высокоэффективная жидкостная хроматография - вариант колоночной жидкостной хроматографии, в которой подвижная фаза элюент - проходит через заполняющий колонки сорбент с большой скоростью за счёт значительного давления на входе в хроматографическую колонку. Поэтому ВЭЖХ позволяет разделять многокомпонентные смеси на индивидуальные вещества высокой степени чистоты. ВЭЖХ отличается высокой чувствительностью. На разделе 10-15 компонентов затрачивается 20-30 мин. Жидкостный хроматограф включает такие узлы ,как дозатор, насос высокого давления высокоэффективная колонка ,детектор с регистрирующим устройством .Колонки изготавливают из нержавеющей стали и плотно набиваются адсорбентом с размером частиц 5-10мкм.В качестве элюента используют различные углеводороды в сочетании с этанолом. Детектором служит спектрофотометр с переменной длиной волны ,но существуют также флуориметрические ,электрохимические и др. детекторы. Подлинность испытуемых ЛВ подтверждают по времени выхода каждого компонента смеси из колонки, которое будет стабильно при одинаковых условиях проведения эксперимента .Количественное содержание рассчитывается по площади пика ,которая пропорциональна количеству ЛВ в пробе.
17.Титрованные растворы. Приготовление и стандартизация титрованных растворов. Вычисление коэффициента поправки методом отдельных навесок и методом пипетирования по ГФ13.
Титрованными растворами называются растворы точно известной концентрации, предназначенные для целей титриметрического анализа. Титр – это выраженная в миллиграммах масса растворенного вещества, содержащаяся в одном миллилитре раствора (размерность – мг/мл). Для приготовления титрованных растворов применяют химически чистые вещества или промышленного производства стандарт-титры (фиксаналы) для титриметрии. Допускается приготовление титрованного раствора приблизительно требуемой концентрации, несколько большей, чем требуется по расчету, который при необходимости можно довести до нужной концентрации путем разведения или укрепления. Приготовленные титрованные растворы стандартизуют двумя способами: по стандартному титрованному раствору или по точной навеске исходного стандартного вещества. Перед стандартизацией титрованный раствор необходимо тщательно перемешать. Концентрацию титрованных растворов определяют путем достаточного количества титрований (не менее трех).
Если титрованный раствор используют в количественном анализе, в котором конечную точку титрования определяют электрометрическим методом (например, методом амперометрии или потенциометрии), раствор стандартизуют тем же методом. Состав среды, в которой стандартизуют титрованный раствор, должен быть таким же, как и тот, в котором он будет использован. Если концентрация приготовленного титрованного раствора отличается от теоретически заданной (например, из-за длительного хранения), то необходимо вычислить поправочный коэффициент (К), представляющий собой отношение фактически полученной концентрации титрованного раствора к теоретически заданной. Вычисление поправочного коэффициента производят одним из указанных ниже способов. Способ 1 – по навеске исходного стандартного вещества:
,где а – навеска вещества, по которому устанавливают титр, г;
Т – количество вещества, по которому устанавливается титр,
содержащееся в 1 мл раствора заданной нормальности, г;
V – объем приготовленного раствора, израсх-ный на Титр-е, мл.
Способ 2 – по титрованному раствору известной концентрации:
,где V0 – объем раствора вещества, по которому устанавливается титр, мл;
V – объем приготовленного раствора, израсходованного на титрование, мл;
K0 – поправочный коэффициент раствора, по которому устанавливается титр.
Титрованные растворы хранят при комнатной температуре, защищая их, при необходимости, от воздействия углерода диоксида и влаги воздуха и от прямых солнечных лучей.
Рекомендуется готовить, стандартизовать и использовать титрованные растворы при одной и той же температуре.
18.ОФС ГФ13: реакции подлинности (идентификации) на катионы: алюминий, аммоний, висмут, железо, калий, кальций, магний, натрий, свинец, серебро, сурьма, цинк. (химизм процесса обязателен).
Алюминий. Около 15 мг лекарственного средства растворяют в 2 мл воды. К полученному раствору или к 2 мл раствора, приготовленного как указано в частной фармакопейной статье, прибавляют 0,5 мл хлористоводородной кислоты разведенной 8,3 % и 0,5 мл реактива тиоацетамида; осадок не образуется. Затем по каплям прибавляют раствор натрия гидроксида разведенный 8,5 %; образуется гелеобразный белый осадок, растворимый при последующем прибавлении раствора натрия гидроксида разведенного 8,5 %. Постепенно прибавляют 10 % раствор аммония хлорида; снова образуется гелеобразный белый осадок. Тиоацетамид в кислой среде гидролизуется с образованием сульфид ионов (вместо токсичного сероводорода).

Соль сульфида алюминия имеет высокую степень растворимости и по- этому осадок не образуется, в отличие от ионов (Be2+, Pb2+, Sn2+, Sn4+ и Sb3+), взаимодействующих с S2- с образованием осадка. Перечисленные ионы как и Al3+ обладают амфотерными свойствами и с гидроксидом натрия образуют гелеобразные осадки. Для исключения ложноположительных реакций и проводится реакция с тиоацетамидом. После прибавления гидроксида натрия образуется осадок: Al3++3OH- →Al(OH)3 ¯ ;
при последующем добавлении гидроксида натрия осадок растворяется:
Al(OH)3 ¯ +OH-(избыток) →[Al(OH)4]-
После добавления к полученному раствору аммония хлорида, вновь выпадает гелеобразный осадок:
[Al(OH)4]-+NH4+ →Al(OH)3 ¯ +NH3+H2O.
Аммоний.1 мл раствора соли аммония (2–6 мг аммоний-иона) нагревают с 0,5 мл 10 % раствора натрия гидроксида; выделяется аммиак, обнаруживаемый по запаху и по посинению влажной красной лакмусовой бумаги. NH4+ + OH- → NH3↑ + H2O
Висмут. А. Указанное в частной фармакопейной статье количество лекарственного средства (около 50 мг висмут-иона) взбалтывают с 3 мл хлористоводородной кислоты разведенной 8,3 % и фильтруют. К фильтрату прибавляют 1 мл 2 % раствора натрия сульфида или сероводорода; образуется коричневато-черный осадок, растворимый при прибавлении равного объема азотной кислоты концентрированной.
1)2BiONO3 + 3Na2S + 4HCI → 2Bi2S3↓ + 2NaNO3 + 4 NaCI + 2H2O 2)2Bi3+ + 3S2- → Bi2S3↓
Железо(II). К 2 мл раствора соли железа(II) (около 20 мг железо(II)-иона) прибавляют 0,5 мл хлористоводородной кислоты разведенной 8,3 % и 1 мл 5 % раствора калия феррицианида; образуется синий осадок. FеСl2 + K3[Fe(CN)6] ® 2КСl + KFeFe(CN)6¯
Железо(III).А. К 2 мл раствора соли железа(III) (около 1 мг железо(III)-иона) прибавляют 0,5 мл хлористоводородной кислоты разведенной 8,3 % и 1–2 капли 5 % раствора калия ферроцианида; образуется синий осадок. Fe3+ + 4К+ + [Fe(CN)6]4- → KFeFe(CN)6¯ + 3К+
Б. К 2 мл раствора соли железа(III) (около 1 мг железо(III)-иона) прибавляют 0,5 мл хлористоводородной кислоты разведенной 8,3 % и 1–2 капли 5 % раствора аммония тиоцианата; появляется красное окрашивание. Fe3+ + 6CNS- → [Fe(CNS)6]3-
В. К раствору соли железа(III) (около 1 мг железо(III)-иона) прибавляют раствор аммония сульфида; образуется черный осадок, растворимый в разведенных минеральных кислотах.
1)2Fe3+ + 3S2- → 2FeS↓ + S 2) Fe2+ + S2- → FeS↓
Калий. А. К 2 мл раствора соли калия (10–20 мг калий-иона) прибавляют 1 мл 20 % раствора винной кислоты, 1 мл 10 % раствора натрия ацетата, 0,5 мл спирта 96 % и встряхивают; постепенно образуется белый кристаллический осадок, растворимый в разведенных минеральных кислотах и растворах гидроксидов щелочных металлов. KCI + H2C4H4O6 → KHC4H4O6↓ + HCI
Б. К 2 мл раствора соли калия (5–10 мг калий-иона), предварительно прокаленной для удаления солей аммония, прибавляют 0,5 мл уксусной кислоты разведенной 30 % и 0,5 мл 10 % раствора натрия кобальтинитрита; образуется желтый кристаллический осадок.
Na3[Co(NO2)6] + 2KCI → K2Na[Co(NO2)6↓ + 2NaCI
В. Соль калия, внесенная в бесцветное пламя, окрашивает его в фиолетовый цвет или при рассматривании через синее стекло – в пурпурно-красный.
Кальций.А. К 1 мл раствора соли кальция (2–20 мг кальций-иона) прибавляют 1 мл 4 % раствора аммония оксалата; образуется белый осадок, нерастворимый в уксусной кислоте разведенной 30 % и 10 % растворе аммиака, растворимый в разведенных минеральных кислотах. C2O42- + Ca2+ ® СаС2О4¯
Б. Соль кальция, смоченная хлористоводородной кислотой 25 % и внесенная в бесцветное пламя, окрашивает его в кирпично-красный цвет.
Магний. К 1 мл раствора соли магния (2–5 мг магний-иона) прибавляют 1 мл 10 % раствора аммония хлорида, 1 мл 10 % раствора аммиака и 0,5 мл 5 % раствора натрия фосфата; образуется белый кристаллический осадок, растворимый в разведенных минеральных кислотах и уксусной кислоте. МgCl2 + Na2HPO4 + NH3 NH4Cl NH4МgPO4¯+ 2 NaCl
Натрий. А. К 2 мл раствора натриевой соли (7–10 мг натрий-иона) прибавляют 2 мл 15 % раствора калия карбоната и нагревают до кипения; осадок не образуется. К раствору прибавляют 4 мл раствора калия пироантимоната и нагревают до кипения. Охлаждают в ледяной воде и при необходимости потирают внутренние стенки пробирки стеклянной палочкой; образуется плотный осадок белого цвета.
Na++K[Sb(OH)6]®Na[Sb(OH)6] ↓ +K+ Б. Соль натрия, смоченная хлористоводородной кислотой 25 % и внесенная в бесцветное пламя, окрашивает его в желтый цвет.
Цинк. А. К 2 мл нейтрального раствора соли цинка (5–20 мг цинк-иона) прибавляют 0,5 мл 2 % раствора натрия сульфида или сероводорода; образуется белый осадок, нерастворимый в уксусной кислоте разведенной 30 % и легко растворимый в хлористоводородной кислоте разведенной 8,3 %. Zn2+ + S2-® ZnS¯
Б. К 2 мл раствора соли цинка (5–20 мг цинк-иона) прибавляют 0,5 мл 5 % раствора калия ферроцианида; образуется белый осадок, нерастворимый в хлористоводородной кислоте разведенной 8,3 %. 3Zn2+ + 2K+ + 2[Fe(CN)6]4- ® K2Zn3[Fe(CN)6]2¯
Серебро.(Аg+)
1.Реакция восстановления Ag+ из аммиачного раствора серебра нитрата до металлического серебра при нагревании с раствором формальдегида (реакция «серебряного зеркала»):
AgNO3 + 2NH4OH →[Ag(NH3)2]NO3 + 2Н2O
t0
2[Ag(NH3)2]NO3 + HCOH + H2O → 2Ag↓ + HCOONH4 + 2NH4NO3 + NH3↑
Металлическое серебро образует на стенках пробирки тонкую блестящую пленку - «серебряное зеркало».
2. Реакция осаждения кислотой хлороводородной или натрия хлоридом. Образуется белый творожистый осадок AgCl:
AgNO3 + HCl → AgCl↓+HNO3
Белый творожистый
Осадок серебра хлорида нерастворим в кислоте азотной разведенной, растворяется в растворе аммиака с образованием комплексного соединения:
AgCl + 2NH4OH→[Ag(NH3)]Cl + 2Н2O
3. Реакция осаждения раствором натрия гидроксида или раствором аммиака:
2AgN03 + 2NH4OH → Ag2O↓ + 2NH4NO3
черный осадок
Осадок серебра оксида нерастворим в избытке натрия гидроксида, растворяется в избытке раствора аммиака с образованием комплексного соединения:
Ag2O + 4NH4OH → 2[Ag(NH3)2]OH +ЗН2O
Свинец.1. Калия йодид образует с солями свинца (ΙΙ) желтый осадок дийодида свинца, растворимый в избытке реактива с образованием бесцветного раствора:

2. Растворы хромата калия и дихромата калия образуют с растворами солей свинца желтый осадок хромата свинца:

23. ОФС в ГФ 13: определение азотосодержащих веществ (первичных, вторичных, третичных аминов) методом нитриметрии. Сущность метода, химизм и особенности проведения титриметрической реакции, индикаторы, пример применения.
Нитритометрия – метод титриметрического анализа, при котором в качестве титрованного раствора используется раствор натрия нитрита. Применяется для количественного определения соединений, содержащих первичную или вторичную ароматическую аминогруппу, для определения гидразидов, а также ароматических нитросоединений после предварительного восстановления нитрогруппы до аминогруппы. Методика. Если не указано иначе, точную навеску образца лекарственного средства, указанную в фармакопейной статье, растворяют в смеси 10 мл воды и 10 мл хлористоводородной кислоты разведенной 8,3 %. Прибавляют воду до общего объема 80 мл, 1 г калия бромида и при постоянном перемешивании титруют натрия нитрита раствором 0,1 М. В начале титрования прибавляют раствор натрия нитрита со скоростью 2 мл/мин, а в конце (за 0,5 мл до эквивалентного количества) – 0,05 мл/мин. Титрование проводят при температуре раствора 15 — 20 ºС, однако в некоторых случаях требуется охлаждение до 0 — 5 ºС. При потенциометрическом титровании в качестве индикаторного применяют платиновый электрод, в качестве электродов сравнения используют хлорсеребряный или насыщенный каломельный электрод. При амперометрическом титровании на электроды накладывают разность потенциалов 0,3 — 0,4 В, если не указано иначе в фармакопейной статье. Точку эквивалентности определяют электрометрическими методами (потенциометрическое титрование, титрование «до полного прекращения тока») или с помощью внутренних индикаторов и внешнего индикатора (йодкрахмальная бумага). В качестве внутренних индикаторов используют тропеолин ОО (4 капли раствора), тропеолин ОО в смеси с метиленовым синим (4 капли раствора тропеолина ОО и две капли раствора метиленового синего), нейтральный красный (две капли в начале и две капли в конце титрования). Титрование с тропеолином ОО проводят до перехода окраски от красной к желтой, со смесью тропеолина ОО с метиленовым синим – от красно-фиолетовой к голубой, с нейтральным красным – от красно-фиолетовой к синей. Выдержку в конце титрования с нейтральным красным увеличивают до 2 мин. Титрование с йодкрахмальной бумагой ведут до тех пор, пока капля титруемого раствора, взятая через 1 мин после прибавления натрия нитрита раствора 0,1 М, не будет немедленно вызывать синее окрашивание на бумаге. В некоторых случаях выдержка может быть увеличена, о чем должно быть указано в фармакопейной статье. Параллельно проводят контрольный опыт.
21.ИК-спектрометрические методы идентификации лекарственных средств. Связь химического строения и спектральных характеристиками веществ. Аппаратурное оформление метода. Подготовка образцов для исследования.
ИК
–спектр химического соединения
является одной из его наиболее важных характеристик.
ИК область спектра занимает диапазон длин волн от границы видимой до
микроволновой области (0,75 до 200 мк). Однако, обычно под ИК областью
подразумевают более узкий интервал от 2,5 до 16 мк. Для характеристики ИК
излучения более часто используют волновые числа (величины обратные длинам волн
– обратные сантиметры, см -1). Так, интервал 2,5-16 мк соответствует 4000-625
см -1 .
Инфракрасные спектры (колебательные спектры) (ИК-спектры) возникают вследствие
поглощения энергии электромагнитного излучения при колебаниях ядер атомов в
молекулах или ионах, которые сопровождаются изменением дипольных моментов, и
представляют собой зависимость пропускания или поглощения от длины волны
(λ) или частоты колебаний (ν). Применение ИК-спектроскопии в
анализе лекарственных средств ИК-спектроскопия используется: -
при установлении структуры новых БАВ получаемых путем химического синтеза или
выделяемых из природных объектов (животное или растительное сырье, продукты
жизнедеятельности микроорганизмов); изучении строения метаболитов; - при
испытании на подлинность лекарственных веществ; определении доброкачественности
лекарственных соединений; - количественном анализе; - контроле
технологического процесса в промышленном производстве фармпрепаратов (полнота
протекания). Идентификация лекарственного вещества может быть проведена
путем сопоставления ИК-спектра исследуемого вещества с аналогичным спектром
его стандартного образца или с рисунком стандартного спектра, приведенного в
фармакопейной статье. ИК-спектр испытуемого образца должен
иметь полное совпадение полос поглощения с полосами поглощения стандартного
спектра по положению и относительной интенсивности. Спектр содержит
некоторый набор полос разной интенсивности, который определяется химическим
составом образца. Каждое вещество обладает своим неповторимым химическим
составом и строением образующих его молекул и, соответственно, своим уникальным
спектром поглощения, который служит так называемым «молекулярным отпечатком»
для данного вещества. Молекула, состоящая из N атомов, имеет 3N степеней
свободы – это число независимых параметров для описания положения всех атомов
молекулы в декартовых координатах (x, y, z). В нелинейной молекуле из
всех 3N независимых параметров три степени свободы приходятся на поступательное
движение молекулы как целого и три – на вращательное движение молекулы вокруг
ее главных осей. Оставшиеся 3N-6 степеней свободы представляют собой так
называемые нормальные колебания – независимые повторяющиеся сами по себе
движения молекулы. Для линейной молекулы характерно 3N-5
нормальных колебаний, так как линейные молекулы имеют три поступательных и две
вращательных степени свободы молекулы как целого. Полное колебательное движение
молекулы можно представить в виде комбинации нормальных колебаний.
Подготовка проб Существуют различные способы подготовки
исследуемого образца в ИК-спектрофотометрии: 1. Растворы веществ В
связи с тем, что в ИК-области поглощает любое вещество, в качестве
растворителей используют соединения простейшей структуры, спектры которых
состоят из минимального числа полос, и наиболее часто – CCl4 и CS2. Для
растворов применяют цилиндрические кюветы толщиной 0,1-1 мм с окнами из солевых
пластин. Необходимый для заполнения кюветы объем раствора 0,1- 1,0 мл при
концентрации 0,05-10%.
2. Тонкие пленки (< 0,01 мм) жидкого вещества, помещенные 55 между
солевыми пластинами, удерживаемыми капиллярными силами. 3. Пасты,
приготовляемые тщательным растиранием твердого образца с вазелиновым маслом и
помещаемые в виде тонкого слоя между солевыми пластинами. Само вазелиновое
масло поглощает в области 2900 и 1400 см-1 Методика получения пасты с
вазелиновым маслом более проста и экспрессна. 4. Твердые вещества в
виде тонкого порошка 0,5-1 г тщательно перемешанные с порошком бромида калия
≈100 мг и затем спрессованные в специальном устройстве под давлением в
тонкую пластину. Приборы для регистрации ИК-спектров. Могут быть
использованы инфракрасные спектрофотометры, снабженные оптической системой
(призмы или дифракционные решетки), выделяющей монохроматическое излучение в
измеряемой области, или спектрофотометры с Фурье-преобразованием. В последних
используется полихроматическое излучение и рассчитывается спектр в заданной
области частот путем Фурье-преобразования исходных данных. В таких приборах
вместо диспергирующего прибора используется интерферометр, а обработка
спектральных данных производится с помощью компьютера.
19.ОФС ГФ13: реакции подлинности (идентификации) на анионы: бромиды, йодиды, карбонаты, гидрокарбонаты, сульфаты, сульфиты, сульфиды, фосфаты, хлориды, силикаты, нитраты, нитриты. ( Химизм процессов обязателен).

Бромиды. А. К 1 мл раствора бромида (2–30 мг бромид-иона) прибавляют 1 мл хлористоводородной кислоты разведенной 8,3 %, 0,5 мл 5 % раствора хлорамина, 1 мл хлороформа и взбалтывают; хлороформный слой окрашивается в желто-бурый цвет.
Б. К 2 мл раствора бромида (2–10 мг бромид-иона) прибавляют 0,5 мл азотной кислоты разведенной 16 % и 0,5 мл 2 % раствора серебра нитрата; образуется желтоватый творожистый осадок, нерастворимый в азотной кислоте разведенной 16 % и трудно растворимый в 10 % растворе аммиака.
NaBr + AgNO3 → AgBr↓ + NaNO3
AgBr + 2NH3. 2H2O → [Ag(NH3)2]Br
Йодиды. А. К 2 мл раствора йодида (3–20 мг йодид-иона) прибавляют 0,2 мл серной кислоты разведенной 16 %, 0,2 мл 10 % раствора натрия нитрита или 3 % раствора железа(III) хлорида и 2 мл хлороформа; при взбалтывании хлороформный слой окрашивается в фиолетовый цвет.
2KI + 2NaNO2 + 2H2SO4 → I2 + 2NO↑ + Na2SO4 + K2SO4 + 2H2O
Б. К 2 мл раствора йодида (2–10 мг йодид-иона) прибавляют 0,5 мл азотной кислоты разведенной 16 % и 0,5 мл 2 % раствора серебра нитрата; образуется желтый творожистый осадок, нерастворимый в азотной кислоте разведенной 16 % и 10 % растворе аммиака.
I- + Ag+ → AgI↓
В. При нагревании 0,1 г лекарственного средства с 1 мл серной кислоты концентрированной выделяются пары фиолетового цвета.
2KI + 2H2SO4 → I2↑ + SO2↑+ K2SO4 + 2H2O
Карбонаты (Гидрокарбонаты). А. К 0,2 г карбоната (гидрокарбоната) или к 2 мл раствора карбоната (гидрокарбоната) (1:10) прибавляют 0,5 мл хлористоводородной кислоты разведенной 8,3 %; выделяется газ, при пропускании которого через раствор кальция гидроксида образуется белый осадок.
NaHCO3 + НСl ® NaCl + CO2 + Н2О
СО2 + Са(ОН)2 ® СаСО3 ¯ + Н2О
Б. К 2 мл раствора карбоната (1:10) прибавляют 5 капель насыщенного раствора магния сульфата; образуется белый осадок (гидрокарбонат образует осадок только при кипячении смеси).
4Na2CO3 + 4MgSO4 + 4Н2О ® 3MgCO3.Mg(OH)2.3H2O¯ + CO2+ 4Na2SO4
В. Раствор карбоната (1:10) при прибавлении 1 капли 1 % раствора фенолфталеина окрашивается в красный цвет (отличие от гидрокарбоната).
СО32- + НOН ® ОН- + НСО3-
НСО3- + НОН ® ОН- + Н2О + CO2
Сульфаты.К 2 мл раствора сульфата (5–50 мг сульфат-иона) прибавляют 0,5 мл 5 % раствора бария хлорида; образуется белый осадок, нерастворимый в разведенных минеральных кислотах.
SO42- + Ba2+ → BaSO4↓
Сульфиты. А. К 2 мл раствора сульфита (10–30 мг сульфит-иона) прибавляют 2 мл хлористоводородной кислоты разведенной 8,3 % и встряхивают; постепенно выделяется сернистый газ, обнаруживаемый по характерному резкому запаху.
SO32- + 2H+ → H2SO3 → SO2↑ + H2O
Б. К 2 мл раствора сульфита (2–20 мг сульфит-иона) прибавляют 0,5 мл 5 % раствора бария хлорида; образуется белый осадок, растворимый в хлористоводородной кислоте разведенной 8,3 % (отличие от сульфатов).
SO32- + Ba2+ → BaSO3↓
BaSO3 + 2HCI → BaCI2 + H2O + SO2↑
Фосфаты. А. К 1 мл раствора фосфата (10–30 мг фосфат-иона), нейтрализованного до рН около 7,0, прибавляют несколько капель 2 % раствора серебра нитрата; образуется желтый осадок, растворимый в азотной кислоте разведенной 16 % и 10 % растворе аммиака.
PO43- + 3Ag+ → Ag3PO4↓
Ag3PO4 ¯ + 6NH4OH →3[Ag(NH3 )2]3+ + PO43-+6H2O
Б. К 1 мл раствора фосфата (10–30 мг фосфат-иона) прибавляют 1 мл 10 % раствора аммония хлорида, 1 мл 10 % раствора аммиака и 0,5 мл 10 % раствора магния сульфата; образуется белый кристаллический осадок, растворимый в разведенных минеральных кислотах.
Na3PO4 + 3NH4CI + NH3. Н2О + МgSO4 ® NH4МgPO4¯+ 3NaCl + (NH4)2SO4
MgNH4PO4¯ +3HCl®MgCl2+NH4Cl+H3PO4
В. К 1 мл раствора фосфата (10–30 мг фосфат-иона) в азотной кислоте разведенной 16 % прибавляют 2 мл 10 % раствора аммония молибдата и нагревают; образуется желтый кристаллический осадок, растворимый в 10 % растворе аммиака.
H3PO4 + 12(NH4)MoO4 + 21HNO3 ® (NH4)3PO4 ·12MoO3¯ + 21NH4NO3 + 12H2O
(NH4)3PO4· 12 MoO3 ¯ + 23NH4OH ® 12(NH4)2 MoO4 + (NH4)2HPO4+11H2O
Хлориды.К 2 мл раствора хлорида (2–10 мг хлорид-иона) прибавляют 0,5 мл азотной кислоты разведенной 16 % и 0,5 мл 2 % раствора серебра нитрата; образуется белый творожистый осадок, нерастворимый в азотной кислоте разведенной 16 % и растворимый в 10 % растворе аммиака. Для солей органических оснований испытание растворимости образовавшегося осадка проводят после отфильтровывания и промывания осадка водой.
Cl- + Ag+ → AgCl↓
AgCl¯ + 2NH4OH ® [Ag(NH3)2]++ Cl- + 2H2O
Нитраты. А. К лекарственному средству (около 1 мг нитрат-иона) прибавляют 2 капли раствора дифениламина; появляется синее окрашивание.
Б. К лекарственному средству (2–5 мг нитрат-иона) прибавляют по 2–3 капли воды и серной кислоты концентрированной, кусочек металлической меди и нагревают; выделяются пары бурого цвета.
2NaNO3 + H2SO4 → Na2SO4+ 2HNO3
Cu + HNO3 → Cu(NO3)2 + NO2↑ + H2O
Нитриты.А. К лекарственному средству (около 1 мг нитрит-иона) прибавляют 2 капли раствора дифениламина; появляется синее окрашивание.
Дифенилдифенохинондииминбензидин (сернокислая соль)

Б. К лекарственному средству (около 30 мг нитрит-иона) прибавляют 1 мл серной кислоты разведенной 16 %; выделяются желто-бурые пары (отличие от нитратов).
NaNO2 + H2SO4 → NaHSO4 + HNO2
2HNO2 → NO↑ + NO2↑ + H2O
В.
Несколько кристаллов антипирина растворяют в фарфоровой чашке в 2 каплях
хлористоводородной кислоты разведенной 8,3 %, прибавляют 2 капли раствора
нитрита (около 1 мг нитрит-иона); появляется зеленое окрашивание (отличие от
нитратов).
Силикаты.
K2SiO3 + H2SO4 = K2SO4 + H2SiO3
2K(+) + SiO3(2-) + 2H(+) + SO4(2-) = 2K(+) + SO4(2-) + H2SiO3
SiO3(2-) + 2H(+) = H2SiO3
20.Реакции подлинности (идентификации) на органические анионы: ацетаты, ацетил, бензоаты, лактаты, силицилаты, тартраты, цитраты.
Ацетаты. А. 2 мл раствора ацетата (20–60 мг ацетат-иона) нагревают с равным количеством серной кислоты концентрированной и 0,5 мл спирта 96 %; появляется характерный запах этилацетата.
CH3COONa + C2H5OH + H2SO4 →CH3COOС2Н5 + NaHSO4 +СО2
Б. К 2 мл нейтрального раствора ацетата (20–60 мг ацетат-иона) прибавляют 0,2 мл 3 % раствора железа(III) хлорида; появляется красно-бурое окрашивание, исчезающее при прибавлении разведенных минеральных кислот.
3CH3COONa + FeCl3 → Fe(CH3COO)3 + 3NaCl
3Fe(CHOO)3 + 2H2O → [Fe3(OH)2(CH3COO)6]CH3COO + 2CH3COOH [Fe3(OH)2(CH3COO)6]CH3COO + 9HCl → 3FeCl3 + 7CH3COOH + 2H2O
Бензоаты. К 2 мл нейтрального раствора бензоата (10–20 мг бензоат-иона) прибавляют 0,2 мл 3 % раствора железа(III) хлорида; образуется розовато-желтый осадок, растворимый в эфире.
6С6H5COO−+2Fe3++10H2O →(С6H5COO)3Fe⋅Fe(OH)3⋅7H2O+3C6H5COOH
Салицилаты. К 2 мл нейтрального раствора салицилата (2–10 мг салицилат-иона) прибавляют 2 капли 3 % раствора железа(III) хлорида; появляется сине-фиолетовое или красно-фиолетовое окрашивание, которое сохраняется при прибавлении небольшого количества уксусной кислоты разведенной 30 %, но исчезает при прибавлении хлористоводородной кислоты разведенной 8,3 %. При этом образуется белый кристаллический осадок.
Тартраты. А. К 1 мл раствора тартрата (около 20 мг тартрат-иона) прибавляют кристаллик калия хлорида, 0,5 мл спирта 96 %; образуется белый кристаллический осадок, растворимый в разведенных минеральных кислотах и растворах гидроксидов щелочных металлов.

Б. 0,25 мл раствора тартрата (около 5 мг тартрат-иона) нагревают с 1 мл серной кислоты концентрированной и несколькими кристаллами резорцина; через 15–30 споявляется вишнево-красное окрашивание.

Цитраты. А. К 1 мл нейтрального раствора цитрата (2–10 мг цитрат-иона) прибавляют 1 мл 20 % раствора кальция хлорида; раствор остается прозрачным; при кипячении образуется белый осадок, растворимый в хлористоводородной кислоте разведенной 8,3 %.

Б. К лекарственному средству (1–2 мг цитрат-иона) прибавляют 0,5 мл уксусного ангидрида и нагревают; через 20–40с появляется красное окрашивание.
При дегидратации лимонной кислоты образуется аконитовая кислота (HOOCCH=C(COOH)CH2COOH), при циклизации и декарбоксилированиикоторой получаются соединения фуранового и пиранового рядов. Носителем окраски, по-видимому, является 6-ацетоксипиранон-2, образующийся в результате этих превращений:

22.ОФС в ГФ 13: определение азота в органических соединениях методом Къельдаля. Химизм метода, аппаратурное оформление, варианты реализации метода. Пример применения.
Метод основан на минерализации лекарственного средства под воздействием серной кислоты концентрированной при нагревании в присутствии катализаторов. В качестве катализаторов возможно использование смеси калия сульфата, меди сульфата и/или селена и/или титана диоксида. При этом азот превращается в аммония сульфат. При прибавлении натрия гидроксида выделяется аммиак, который перегоняют с паром в приемник, содержащий кислоту для его поглощения: борную – в методе прямого титрования (1 и 2); серную или хлористоводородную – в методе обратного титрования (3). В методах 1 и 2 поглощенный аммиак титруют раствором хлористоводородной или серной кислоты, в методе 3 избыток кислоты оттитровывают раствором натрия гидроксида. По результатам титрования рассчитывают содержание азота. Различают следующие варианты метода: 1) метод Къельдаля, 2) микрометод Къельдаля, 3) метод Къельдаля (обратное титрование).
Прибор для определения азота (рисунок) состоит из парообразователя – круглодонной колбы (1) вместимостью 3 л с предохранительной трубкой (2), сменных колб Къельдаля с длинным горлом (3) для конденсации водяных паров и защиты от потери вещества, воронки (4) с зажимом или краном (5) для прибавления натрия гидроксида, брызгоуловителя (6), прямого холодильника (7) и сменных конических колб-приемников (8). Стеклянная посуда должна быть термостойкой. Работу на приборе осуществляют в вытяжном шкафу. Вместо описанного прибора могут быть использованы установки для автоматического определения азота по Къельдалю, определение проводят потенциометрически.
Метод Къельдаля В колбу Къельдаля (3) вместимостью 200 – 300 мл (другие объемы от 50 до 500 мл должны быть указаны в фармакопейной статье) помещают точную навеску (указывают в фармакопейной статье) или точный объем образца лекарственного средства (0,5 – 10,0 мл) с содержанием азота около 14 – 35 мг, три стеклянных шарика для пенящихся веществ и 1 г растертой смеси калия сульфата и меди сульфата, взятых в соотношении 10 : 1. Для трудносжигаемых веществ дополнительно в колбу (3) прибавляют 0,05 г металлического селена и/или 1 мл концентрированного раствора водорода пероксида. Прибавляют 7 мл серной кислоты концентрированной и осторожно вращают колбу для стекания кислоты со стенок и ее перемешивания с содержимым колбы. Постепенно нагревают колбу (3), закрытую стеклянной воронкой, на электронагревательном приборе и далее кипятят содержимое в течение нескольких часов до получения раствора светло-зеленого цвета. На стенках колбы не должно оставаться обугленного вещества. Кипячение продолжают еще 30 мин или более до просветления раствора. Если при кипячении происходит сильное пенообразование, то рекомендуется снять колбу Къельдаля с нагревательного прибора и дать пене осесть, затем снова продолжают нагревание, не допуская попадания пены в горло колбы. После охлаждения колбы Къельдаля в нее осторожно прибавляют 20 мл воды, вращая колбу для перемешивания содержимого, вновь охлаждают и присоединяют колбу к собранному прибору для определения азота (рисунок), заранее промытому путем пропускания через него пара. В парообразователь наливают воду, не менее половины объема, подкисленную 0,5 М или 0,05 М раствором серной кислоты по индикатору метиловому красному (2 – 3 капли) до слабо-розового цвета, для связывания аммиака, который может попасть из воздуха. Для обеспечения равномерного кипения воды в парообразователь помещают стеклянные шарики. В приемник перед началом отгонки наливают 20 мл борной кислоты раствора 4 % и прибавляют 0,25 мл (5 капель) смешанного индикатора. Нижний конец внутренней трубки холодильника должен быть опущен в раствор, находящийся в приемнике. После сборки прибора в холодильник пускают воду и доводят до кипения воду в парообразователе. Затем в колбу (3) из воронки медленно по каплям прибавляют 40 мл натрия гидроксида раствора 30 %, следя за тем, чтобы раствор в колбе (3) энергично перемешивался поступающим паром. Для обеспечения большей герметичности прибора в воронке следует оставлять некоторый избыток натрия гидроксида раствора 30 %. Собирают около 100 мл отгона (или количество, указанное в фармакопейной статье). Во время отгонки колбу Къельдаля нагревают так, чтобы объем жидкости в ней оставался постоянным. По окончании отгонки приемник опускают таким образом, чтобы трубка холодильника находилась над поверхностью жидкости, находящейся в приемнике. Трубку холодильника промывают снаружи водой, продолжая подачу пара в колбу (3) в течение 1 – 2 мин; промывную воду собирают в тот же приемник. После этого прекращают нагревание парообразователя и немедленно отсоединяют колбу Кьельдаля от прибора. По окончании отгонки дистиллят титруют хлористоводородной кислоты раствором 0,1 М или серной кислоты раствором 0,05 М (должно быть указано в фармакопейной статье) до перехода окраски смешанного индикатора из зеленой в красно-фиолетовую.Проводят контрольный опыт таким же образом и с теми же реактивами, но без испытуемого образца; полученный результат используют для внесения поправки при расчете содержания азота.
 Метод Къельдаля
(обратное титрование) А. Б. Данный
метод применяют преимущественно в препаратах крови. В колбу Къельдаля
с помощью калиброванной пипетки вносят 0,5 – 1,0 мл испытуемого раствора,
содержащего 8 – 32 мг азота (или 50 – 200 мг белка), затем вносят
растертую смесь (около 1 г) калия сульфата и меди сульфата (3:1) и от 2 до 4 мл
серной кислоты концентрированной в зависимости от содержания белка. Содержимое
колбы кипятят. Для ускорения сжигания несколько раз прибавляют по 5 капель
концентрированного раствора водорода пероксида и продолжают кипятить, пока
раствор не станет голубым или бесцветным. По окончании сжигания смесь
охлаждают. В парообразователь наливают воду, нагревают до кипения и проверяют
аппарат на герметичность. Присоединяют колбу Къельдаля к прибору для
определения азота или количественно переносят содержимое колбы в реакционный
сосуд прибора, используя 30 мл воды. Отгон собирают в приемник, куда
предварительно наливают от 10,0 до 25,0 мл серной кислоты раствора 0,05 М в
зависимости от содержания белка. Приемник присоединяют так, чтобы нижний конец
трубки холодильника был опущен в раствор. Воду в парообразователе нагревают до
кипения. В колбу Къельдаля или в реакционный сосуд прибора с минерализатом
через воронку прибавляют около 20 мл натрия гидроксида раствора 15 М до
появления коричневой окраски минерализата. После этого быстро и герметично
закрывают зажим воронки и отгоняют аммиак в течение 5 мин. Затем опускают
приемник с серной кислоты раствором 0,05 М так, чтобы нижний конец трубки
холодильника не касался поверхности жидкости, и продолжают перегонку еще 5 мин.
Отгон титруют натрия гидроксида раствором 0,1 М до перехода сиреневой окраски в
зеленую (индикатор – 0,05 мл смешанного индикатора). Параллельно проводят
контрольный опыт. 1 мл натрия гидроксида раствора 0,1 М соответствует
1,401 мг азота.
Метод Къельдаля
(обратное титрование) А. Б. Данный
метод применяют преимущественно в препаратах крови. В колбу Къельдаля
с помощью калиброванной пипетки вносят 0,5 – 1,0 мл испытуемого раствора,
содержащего 8 – 32 мг азота (или 50 – 200 мг белка), затем вносят
растертую смесь (около 1 г) калия сульфата и меди сульфата (3:1) и от 2 до 4 мл
серной кислоты концентрированной в зависимости от содержания белка. Содержимое
колбы кипятят. Для ускорения сжигания несколько раз прибавляют по 5 капель
концентрированного раствора водорода пероксида и продолжают кипятить, пока
раствор не станет голубым или бесцветным. По окончании сжигания смесь
охлаждают. В парообразователь наливают воду, нагревают до кипения и проверяют
аппарат на герметичность. Присоединяют колбу Къельдаля к прибору для
определения азота или количественно переносят содержимое колбы в реакционный
сосуд прибора, используя 30 мл воды. Отгон собирают в приемник, куда
предварительно наливают от 10,0 до 25,0 мл серной кислоты раствора 0,05 М в
зависимости от содержания белка. Приемник присоединяют так, чтобы нижний конец
трубки холодильника был опущен в раствор. Воду в парообразователе нагревают до
кипения. В колбу Къельдаля или в реакционный сосуд прибора с минерализатом
через воронку прибавляют около 20 мл натрия гидроксида раствора 15 М до
появления коричневой окраски минерализата. После этого быстро и герметично
закрывают зажим воронки и отгоняют аммиак в течение 5 мин. Затем опускают
приемник с серной кислоты раствором 0,05 М так, чтобы нижний конец трубки
холодильника не касался поверхности жидкости, и продолжают перегонку еще 5 мин.
Отгон титруют натрия гидроксида раствором 0,1 М до перехода сиреневой окраски в
зеленую (индикатор – 0,05 мл смешанного индикатора). Параллельно проводят
контрольный опыт. 1 мл натрия гидроксида раствора 0,1 М соответствует
1,401 мг азота.
Рисунок – Прибор для определения азота.
1 – парообразователь; 2 – предохранительная трубка; 3 – колба Къельдаля; 4 – воронка; 5 – кран или зажим; 6 – брызгоуловитель; 7 – холодильник; 8 – колба-приемник
24.Определение воды в лекарственных препаратах по ГФ13: потеря массы при высушивании, метод Фишера, метод дистилляции (Дина и Старка).
Метод К. Фишера
(полумикрометод)
Метод основан на химическом
взаимодействии воды с компонентами реактива К. Фишера. Реактив К. Фишера
представляет собой раствор серы диоксида, йода и пиридина (или другого
основания, например, имидазола) в метаноле. Взаимодействие реактива с водой
протекает в две стадии стехиометрически по уравнениям:
H2O + SO2 + I2 + 3C5H5N®2C5H5N · HI +
C5H5NSO3
C5H5NSO3 + CH3OH ®HSO4CH3× C5H5N
Используемые растворы и реактивы должны быть безводными. Их хранят и применяют
в условиях, исключающих возможность воздействия на них атмосферной влаги.
Йодсернистый реактив представляет собой раствор, содержащий пиридин
безводный, монометиловый эфир этиленгликоля, йод и серу диоксид Использование
реактивов такого состава должно быть предварительно валидировано для
подтверждения в каждом конкретном случае стехиометрии реакции и отсутствия
несовместимости между испытуемым веществом и реактивом. При определении воды в
твердых веществах, нерастворимых в метаноле, тонко измельченную навеску
вещества взбалтывают с метанолом, после чего титруют реактивом К. Фишера. Пропанол
и другие алканолы имеют большую растворяющую способность для молекул с
длинной цепью и могут использоваться как таковые или в смеси с метанолом при
анализе высокомолекулярных соединений. 2-Метоксиэтанол (монометиловый эфир
этиленгликоля) применяют в тех случаях, когда в присутствии метанола
протекают побочные реакции (этерификация, образование кеталей и т. п.).. Хлороформ
является хорошим растворителем для жиров и может использоваться в смеси с
метанолом, содержание которого обычно составляет 50 %, но не менее 25 %. Формамид
улучшает растворимость полярных веществ и может добавляться в метанол для
определения воды в протеинах. Не рекомендуется использование в качестве рабочей
среды чистые апротонные растворители, которые нарушают стехиометрию реакции К.
Фишера. Масса навески, время взбалтывания навески с растворителем, а также
наименование растворителя, должны быть указаны в фармакопейной статье. С
помощью реактива К. Фишера может быть определена как гигроскопическая, так и
кристаллизационная вода. При этом воду можно определять в органических и неорганических
соединениях, в различных растворителях и летучих веществах.
Прибор для титрования по методу К. Фишера представляет собой закрытую
систему, состоящую из бюретки, снабженной осушительной трубкой, заполненной,
осушающим агентом, (например, молекулярными ситами), сосуда для подачи реактива
и колбы для титрования, соединенных с бюреткой. Колба для титрования
представляет собой сосуд вместимостью 60–100 мл с двумя платиновыми
электродами, трубкой для подвода азота, 3 осушительной трубкой, заполненной, осушающим
агентом, (например, молекулярными ситами), и пробкой, в которую вставляется
кончик бюретки. Испытуемое вещество вносят в сосуд через трубку, расположенную
с противоположной стороны по отношению к трубке-осушителю, и закрываемую
притертой пробкой. Перемешивание раствора в процессе титрования осуществляют
при помощи магнитной мешалки или продуванием высушенного азота через раствор.
Конечную точку титрования определяют амперометрически. Электрическая
схема состоит из потенциометра с сопротивлением 2000 Ом, подключенного к
источнику постоянного тока с напряжением 1,5 В и обеспечивающего необходимую
разность потенциалов. Конечную точку титрования допускается определять
визуально по изменению окраски титруемой жидкости от желтой до
красновато-коричневой при условии обеспечения необходимой точности. При этом
необходимо проводить контрольный опыт. Допускается использование автоматических
титраторов в соответствии с инструкцией производителя. Если нет других указаний
в фармакопейной статье, используют методику
Методика А. Точную навеску испытуемого вещества, содержащую
приблизительно от 30 до 50 мг воды, помещают в сосуд для титрования, в который
предварительно внесено 5,0 мл метанола безводного. Перемешивают 1 мин и титруют
реактивом К. Фишера, прибавляя его при приближении к конечной точке по 0,1–0,05
мл. 4 Параллельно проводят контрольный опыт (титруют 5,0 мл метанола
безводного).
Определение воды методом
дистилляции
Прибор.
Определение проводят в приборе (рис. 1), состоящем из стеклянной круглодонной
колбы (1) вместимостью от 250 до 500 мл, приемника (2), представляющего собой
градуированную пробирку или бюретку вместимостью 6–10 мл с ценой деления 0,1
мл, и холодильника (3).
Методика
В колбу (1) отвешивают с точностью до 1 % указанное в фармакопейной статье
количество испытуемого вещества (от 10,0 до 20,0 г (точная навеска), содержащее
от 2 до 3 мл воды), прибавляют 100 мл толуола или Рис. 1. Прибор для
определения воды методом дистилляции 1 – колба, 2 – приемник, 3 – холодильник 8
ксилола и несколько кусочков пористого материала (например, несколько кусочков
пемзы). Колбу нагревают на электроплитке или песчаной бане до кипения.
Кипячение ведут так, чтобы конденсирующийся растворитель не скапливался в
холодильнике, а спокойно стекал навстречу поднимающимся парам жидкости со
скоростью от 2 до 4 капель в секунду. Кипячение прекращают, когда объем воды в
приемнике перестанет увеличиваться и верхний слой растворителя в приемнике
станет прозрачным. Внутреннюю трубку холодильника промывают толуолом и
продолжают нагревание еще 5 мин, после чего приемник охлаждают до комнатной
температуры и стряхивают со стенок приемника все капли воды. Вся отогнанная
вода собирается в нижней части приемника. После полного разделения слоев
отмечают объем отогнанной воды.
25. Определение плотности жидкостей и твердых веществ пикнометрическим методом по ГФ13. Оценочное определение плотности жидкостей с помощью ареометра.
Определение плотности проводят с помощью пикнометра, ареометра или плотномера.
Метод 1 Применяют для определения плотности жидкостей с точностью до ± 0,001 г/cм3 с помощью пикнометра. Чистый сухой пикнометр взвешивают с точностью до 0,0002 г, заполняют с помощью маленькой воронки водой очищенной немного выше метки, закрывают пробкой и выдерживают в течение 20 мин в термостате при температуре (20 ± 0,1) ºС. При этой температуре уровень воды в пикнометре доводят до метки, отбирая излишек воды при помощи пипетки или свернутой в трубку полоски фильтровальной бумаги. Пикнометр снова закрывают пробкой и выдерживают в термостате еще 10 мин. Затем пикнометр вынимают из термостата, проверяют положение мениска воды, который должен находиться на уровне метки. Вытирают фильтровальной бумагой внутреннюю поверхность горлышка и весь пикнометр снаружи, закрывают пробкой. Выдерживают пикнометр под стеклом аналитических весов в течение 10 мин и взвешивают с той же точностью. Пикнометр освобождают от воды, высушивают, ополаскивая последовательно спиртом и эфиром (сушить пикнометр нагреванием не допускается), удаляют остатки эфира продуванием воздуха, заполняют пикнометр испытуемой жидкостью и проводят те же операции, что и с водой. Плотность r20 (г/см3) вычисляют по формуле:
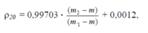 Где :m – пустого пикнометра, г; m1 –пикн с
водой очищ, г; m2 – пикн с испыт жидк-ю, г; 0,99703 –
значение плотности воды при 20 °С, г/см3 (с учетом плотности воздуха);
Где :m – пустого пикнометра, г; m1 –пикн с
водой очищ, г; m2 – пикн с испыт жидк-ю, г; 0,99703 –
значение плотности воды при 20 °С, г/см3 (с учетом плотности воздуха);
0,0012 – значение плотности воздуха при 20 °С и барометрическом давлении 101,1 кПа (760 мм рт. ст.).
 Метод 2 Применяют для определения плотности жидкостей с
точностью до ± 0,001 г/cм3 с помощью пикнометра. Проводят
все операции с водой очищенной и высушивают пикнометр, как описано в методе 1.
При помощи пипетки или небольшой воронки с оттянутым концом вносят в пикнометр
расплавленный жир или воск в таком количестве, чтобы он занимал 1/3 – 1/2
объема пикнометра. Пикнометр без пробки ставят на 1 ч в горячую воду, затем
охлаждают до температуры 20 °С и взвешивают. Содержимое пикнометра доводят
до метки водой очищенной при температуре 20 °С, вытирают пикнометр и снова
взвешивают. В обеих фазах и на поверхности их раздела не должно быть пузырьков
воздуха.
Метод 2 Применяют для определения плотности жидкостей с
точностью до ± 0,001 г/cм3 с помощью пикнометра. Проводят
все операции с водой очищенной и высушивают пикнометр, как описано в методе 1.
При помощи пипетки или небольшой воронки с оттянутым концом вносят в пикнометр
расплавленный жир или воск в таком количестве, чтобы он занимал 1/3 – 1/2
объема пикнометра. Пикнометр без пробки ставят на 1 ч в горячую воду, затем
охлаждают до температуры 20 °С и взвешивают. Содержимое пикнометра доводят
до метки водой очищенной при температуре 20 °С, вытирают пикнометр и снова
взвешивают. В обеих фазах и на поверхности их раздела не должно быть пузырьков
воздуха.
Где m – масса пустого пикнометра, г; m1 – масса пикнометра с водой очищенной, г; m2 – масса пикнометра с жиром, г; m3 – масса пикнометра с жиром и водой, г.
Метод 3 Применяют для определения плотности жидкостей с точностью до ± 0,01 г/см3 с помощью ареометра.Испытуемую жидкость помещают в цилиндр и при температуре 20 ºС осторожно опускают в нее чистый сухой ареометр, на шкале которого предусмотрена ожидаемая величина плотности. Ареометр не должен касаться стенок и дна цилиндра. Через 3 – 4 мин после погружения ареометра производят отсчет по делению шкалы ареометра, соответствующему нижнему мениску жидкости (глаз должен быть на уровне мениска). Примечания. 1.Определение плотности сильно летучих веществ ареометром не допускается. В случае определения плотности темноокрашенных жидкостей отсчет производят по верхнему мениску.
Лекарственные средства неорганической природы.
1.Вода очищенная и вода для инъекций. Способы очистки, контроль качества, хранение.
Вода очищенная
Вода очищенная служит для изготовления перечня жидких лекарственных препаратов и является основой, из которой приготовляют воду для инъекций.
Основные показатели качества:
-pH от 5,0 до 7,0.
-Содержание хлоридов, сульфатов, нитратов, восстанавливающих веществ, кальция, диоксида углерода, тяжелых металлов — отсутствие.
-Содержание аммиака — 0,00002% (в препарате) или не более 0,05 мг/л.
-Микробиологическая чистота — не более 100 микроорганизмов в 1 мл.
-Бесцветность, прозрачность, без вкуса и запаха.
Метод предварительной очистки воды зависит от характера и содержания загрязняющих примесей:
1)Отстаивание, кипячение — для отделения летучих веществ. 2)Отстаивание, фильтрование — удаление механических примесей и взвешенных веществ. 3)Реагентное удаление аммиака. 4)Кипячение или обработка раствором гидроксида кальция — для снижения временной (карбонатной) жесткости воды. 5)Удаление органических веществ обработкой раствором перманганата калия.
Хранение и распределениеВода очищенная хранится и распределяется в условиях, предотвращающих рост микроорганизмов и исключающих возможность любой другой контаминации. Хранение воды очищенной осуществляют в специальных
сборниках, оно не должно превышать 3 сут.
Хранение воды очищенной осуществляют в специальных
сборниках, оно не должно превышать 3 сут.
3.Иод. Получение, контроль качества, условия хранения.
Источниками получения иода служат буровые воды и морские водоросли (в золе последних до 0,5% иода).. Разработкой способа получения иода из морских водорослей занимались в годы первой мировой войны русские ученые Л.В. Писаржевский, и др. О. Ю. Магидсон с сотрудниками (1924–1926 гг) разработал оригинальную технологию получения иода из буровых вод, содержащих 0,001-0,01% иода в виде иодидов. Процесс состоял из ряда последовательных стадий: очистки буровых вод от примеси нефти и нафтеновых кислот, отстаивания от механических примесей, окисления иодид-ионов до свободного иода нитритом натрия в присутствии серной кислоты:
2NaI + 2NaNO2 + 2H2SO4àI2 + 2NO + 2H2O + 2Na2SO4
Выделившийся иод адсорбируют активированным углем. Это наиболее важный этап производства, так как происходит концентрирование (в 200–300 раз) малых количеств иода. Затем иод подвергают десорбции с помощью раствора гидроксида натрия или сульфита натрия:
3I2 + 6NaOH à 5NaI + NaIO3 + 3H2O
I2 + Na2SO3 + H2Oà2HI + Na2SO4
Следующий этап — окисление иодидов до свободного иода с помощью различных окислителей. Наиболее часто используют хлор:
2NaI + Cl2 à I2 + 2NaCl
Процесс окисления может быть осуществлен электролизом. Заключительный этап — процесс очистки иода от примесей. Для этого иод-сырец подвергают сублимации в стальных, чугунных или керамических ретортах.
Iode (Iodum)— Иод -Серовато-черные с металлическим блеском пластинки или сростки кристаллов с характерным запахом.
В водных растворах иодидов иод растворяется с образованием комплексной соли (полииодида):
KI + I2 à K[I3]
Идентифицировать иод можно по окраске его растворов в различных растворителях. Растворы в кислородсодержащих растворителях (вода, эфир) имеют темно-бурую окраску, а в бескислородных (хлороформ) — фиолетовую.
 Подлинность иода
и его лекарственных форм устанавливают с помощью специфической реакции. Она
основана на образовании продукта синего цвета при взаимодействии иода и
крахмального клейстера. С помощью рентгеноструктурного анализа и других
физико-химических методов установлено, что синий иодид крахмала представляет
собой соединения включения (клатраты).. При получении иода из озоленных морских
водорослей или буровых вод может образоваться очень токсичная примесь цианида
иода: I2 + 2C + N2 à 2ICN
Подлинность иода
и его лекарственных форм устанавливают с помощью специфической реакции. Она
основана на образовании продукта синего цвета при взаимодействии иода и
крахмального клейстера. С помощью рентгеноструктурного анализа и других
физико-химических методов установлено, что синий иодид крахмала представляет
собой соединения включения (клатраты).. При получении иода из озоленных морских
водорослей или буровых вод может образоваться очень токсичная примесь цианида
иода: I2 + 2C + N2 à 2ICN
Для установления этой примеси иод обесцвечивают, восстанавливая раствором сернистой кислоты и обнаруживают цианид-ион по образованию берлинской лазури [гексацианоферрат (II) железа (III) натрия], имеющей темно-синюю окраску:
I2 + H2O + H2SO3 à 2HI + H2SO4
ICN + 2NaOHàNaCN + NaIO + H2O
2NaCN + FeSO4 àFe(CN)2 + Na2SO4
Fe(CN)2 + 4NaCN à Na4[Fe(CN)6]
Na4[Fe(CN)6] + FeCl3 à FeNa[Fe(CN)6] + 3NaCl
Количественно иод определяют титрованием 0,1 М раствором тиосульфата натрия в присутствии индикатора — раствора крахмала. Навеску иода предварительно растворяют в водном растворе иодида калия.: I2 + 2Na2S2O3à2NaI + Na2S4O6 . Поскольку процесс протекает в присутствии иодида калия, иод образует вначале комплексное соединение, которое затем взаимодействует с тиосульфатом натрия: I2 + KI à K[I3] , ))) K[I3] + 2Na2S2O3 àKI + 2NaI + Na2S4O6
Кристаллический иод и его 5%-ный раствор хранят с предосторожностью (список Б) в стеклянных банках с притертыми пробками в сухом, прохладном, защищенном от света месте. Иод в медицинской практике применяют в качестве антисептического средства. Иод ядовит, его пары раздражают слизистые оболочки. Хранение В хорошо укупоренной упаковке, в защищенном от света месте.
2.Водорода пероксид. Растворы водорода пероксида. Способы получения, физические и химические свойства, стабильность, контроль качества, хранение и медицинское применение.
В медицинской практике применяют раствор водорода перекиси, магния перекись и гидроперит. По физическим свойствам различают жидкие (3%-ный раствор) и твердые (магния перекись, гидроперит) лекарственные препараты водорода перекиси.
Свойства лекарственных препаратов водорода перекиси
Раствор водорода перекиси H2O2-Бесцветная прозрачная жидкость без запаха3% H2O2
Магния перекисьMgO2 + MgO--Белый легкий порошок, практически нерастворимый в воде25% MgO2. Гидроперит- белый кристаллический порошок, легко растворимый в воде, растворимый в этаноле, практически нерастворимый в хлороформе. Гидроперит образует водорода перекись при растворении в воде. Магния перекись выделяет водорода перекись в расворах минеральных кислот: MgO2 + 2HCl ® MgCl2 + H2O2
Водорода перекись впервые получена Тенаром в 1818 г. при действии серной кислотой на бария перекись: BaO2 + H2SO4 ® H2O2 + BaSO4¯
Производство водорода перекиси осуществляют электролизом 40-68%-ных растворов серной кислоты при 5-8°С. Процесс электролиза проходит по схеме:
 Продуктом электролиза является надсерная
(пероксодисерная) кислота.:
Продуктом электролиза является надсерная
(пероксодисерная) кислота.:
Эти способы позволяют получать разбав-ные р-ры водорода перекиси. Путем перегонки в вакууме 70°С конц-ют водорода перекись, получая в рез-те 30-60%-ные р-ры.
 В химическом отношении водорода перекись представляет
собой очень слабую кислоту. Водные растворы ее имей слабокислую реакцию,
константа диссоциации 2,0.10-12. Водорода перекись
проявляет как окислительные, так и восстановительные свойства. Она устойчива в
чистом состоянии и в водных растворах (при обычной температуре).:
В химическом отношении водорода перекись представляет
собой очень слабую кислоту. Водные растворы ее имей слабокислую реакцию,
константа диссоциации 2,0.10-12. Водорода перекись
проявляет как окислительные, так и восстановительные свойства. Она устойчива в
чистом состоянии и в водных растворах (при обычной температуре).:


Окислительные свойства водорода перекиси используют как для подтверждения подлинности, так и количественного определения. При установлении подлинности к 2 мл раствора водорода перекиси добавляют по I мл разведенной серной кислоты и йодида калия, а затем 5 мл хлороформа. Смесь взбалтывают, после расслоения образовавшийся в результате реакции свободный йод окрашивает слой хлороформа в фиолетовый цвет: Для установления подлинности указанных лекарственных препаратов водорода перекиси используют реакцию образования окрашенных в синий цвет перекисных соединений (смеси надхромовых кислот и пероксида хрома), растворимых в эфире. К раствору водорода перекиси, подкисленному серной кислотой, прибавляют диэтиловый эфир и несколько капель раствора дихромата калия. После взбалтывания и отстаивания смеси эфирный слой окрашивается в синий цвет:


 При выполнении реакции с водорода перекисью высокой
концентрации образуется надхромовая кислота состав Н2Сr2O12
При выполнении реакции с водорода перекисью высокой
концентрации образуется надхромовая кислота состав Н2Сr2O12
Наличие иона магния в магния перекиси подтверждают по образованию белого кристаллического осадка фосфата магния-аммония при взаимодействии с раствором гидрофосфата динатрия в присутствии хлорида аммония и аммиака:

Количе опред-е перекиси вод- восстан вып-ют прямым перманганат-им титр-ем в кислой среде (до слабо-розового окрашивания): Лекарственный препарат должен содержать 2,5-3,5% водорода перекиси. Количественное содержание можно также установить, используя окислительные свойства водорода перекиси, йодометрическим методом:
 Количественное определение магния перекиси и водорода
перекиси в гидроперите проводят прямым перманганатометрическим титрованием.
Они должны соответственно содержать 25% магния перекиси и 35% водорода перекиси
в гидроперите.
Количественное определение магния перекиси и водорода
перекиси в гидроперите проводят прямым перманганатометрическим титрованием.
Они должны соответственно содержать 25% магния перекиси и 35% водорода перекиси
в гидроперите.
Хранят 3%-ный раствор водорода перекиси в склянках с притертыми стеклянными пробками, в прохладном, защищенном от света месте. Твердые препараты водорода перекиси хранят в сухом, защищенном от света месте, в хорошо укупоренной таре при комнатной температуре.
Раствор водорода перекиси применяют в качестве антисептического, дезодорирующего и депигментирующего средства.
4.Иодиды щелочных металлов: натрия иодид и калия иодид. Получение, контроль качества, условия хранения.
Натрия иодид получают при взаимодействии иода и гидроксида натрия с последующим восстановлением иодата натрия сероводородом или пероксидом водорода:
3I2 + 6NaOH à 5NaI + NaIO3 + 3H2O
4NaIO3 + 3H2S + 6NaOH à 4NaI + 3Na2SO4 + 6H2O
Натрия иодид--NaI -Белый кристаллический порошок без запаха, соленого вкуса. Гигроскопичен Калия иодид-KI-Бесцветные или белые кубические кристаллы или белый мелкокристаллический порошок без запаха, солено-горького вкуса. Гигроскопичен
Иодид-ион окисляют раствором нитрита натрия или другим окислителем в кислой среде:
2NaNO2 + 2NaI + 2H2SO4 à I2 + 2NO + 2Na2SO4 + 2H2O
Выделившийся иод окрашивает слой хлороформа в фиолетовый цвет Иодиды под действием концентрированной серной кислоты выделяют фиолетовые пары иода. Из раствора иодидов при добавлении ацетата свинца выпадает осадок иодида свинца желтого цвета:
2NaI + (CH3COO)2Pb à PbI2¯ + 2CH3COONa
Иодиды определяют методом Фаянса в уксуснокислой среде, используя в качестве титранта 0,1 М раствор нитрата серебра и адсорбционный индикатор — эозинат натрия. После осаждения иодид-ионов образующиеся коллоидные частицы иодида серебра от добавления избытка ионов серебра становятся положительно заряженными:
Ag+
[AgI · I–] à [AgI · Ag+]
Описан способ, основанный на окислении иодидов нитритом натрия (калия), позволяющий выполнять определение в присутствии хлоридов, бромидов и различных восстановителей:
2I– + 2NO2– + 4H+ à I2 + 2NO + 2H2O
Натрия и калия хлориды хранят в сухом месте в плотно укупоренных банках. Бромиды и иодиды, кроме того, предохраняют от действия света (в склянках оранжевого цвета).
5.Бромиды щелочных металлов: натрия бромид и калия бромид. Получение, контроль качества, условия хранения.
Подлинность Бромиды: 1. Реакция с нитратом серебра (ГФ
Х). Выпадение желт садка бромида серебра, нерастворимого в NH4OH(10%): NaBr + AgNO3  AgBr↓ + NaNO3,
AgBr↓ + NaNO3,
AgBr + 2NH4OH(10%) →/, AgBr + 2NH4OH(25%) → [Ag(NH3)2]Br + 2H2O.
2. Взаимодействие с окислителями. Слой хлороформа окрашивается в желто-бурый цвет за счет экстракции брома: 2NaBr + 2NaNO2 + 2H2SO4 → Br2 + 2NO↑ + 2Na2SO4 + 2H2O.
Получение. Существуют различные способы промышленного получения бромидов. Один из них основан на использовании железа (II), (III) бромида, который является отходом некоторых химических производств, или получается при обработке железа бромом: Fe + Br2 à FeBr2
3FeBr2 + Br2 àFe3Br8 (FeBr2 . 2FeBr3)
Бромисто-бромное железо нагревают с раствором соды:
toC
Fe3Br8 + 4Na2CO3 + 4H2O à 8NaBr + Fe(OH)2+ 2Fe(OH)3+ 4CO2
(K2CO3) (KBr)
Свойства. Гранулированные порошки белого цвета или мелкие, бесцветные, прозрачные или матовые кристаллы. Легко растворимы в воде, растворимы в 96% спирте. Калия бромид легко растворим в глицерине
Идентификация. 1. Дают реакции соответственно на K+ и Na+ (ГФУ).
2. Реакции на Br-(а,b) (ГФУ). 3. Реакция с сульфатом меди в присутствии конц. H2SO4: Cu2+ + 2Br- à CuBr2
Образующийся черный осадок разрушается при добавлении воды.
Испытания на чистоту.
Броматы: определяют по реакции с калия йодидом в кислой среде, в присутствии крахмала; не должно появляться синего или фиолетового окрашивания:
BrO3- + 5Br- + 6H+ à 3Br2 + 3H2O
2KI + Br2 àI2 + 2KBr
Количественное определение. Аргентометрия по Фольгарду в присутствии дибутилфталата (s=1), пересчет на сухое вещество (ГФУ). Применение. Успокаивающие средства. Хранение. В воздухонепроницаемом контейнере.
6.Хлориды щелочных металлов: натрия хлорид и калия хлорид. Получение, контроль качества, условия хранения.
Натрия хлорид получают из минерала галита, а также из подземных рассолов, воды озер и морей выпариванием. Вначале раствором хлорида бария осаждают сульфаты и фосфаты:
Na2SO4 + BaCl2àBaSO4¯ + 2NaCl
Na2HPO4 + BaCl2 à BaHPO4¯ + 2NaCl
Раствор натрия хлорида отделяют от осадка декантацией, нагревают и обрабатывают избытком карбоната натрия для осаждения примесей солей магния, кальция и бария:
MgCl2 + Na2CO3 à MgCO3¯ + 2NaCl
CaCl2 + Na2CO3 à CaCO3¯ + 2NaCl
BaCl2 + Na2CO3 à BaCO3¯ + 2NaCl
Раствор вновь декантируют и нейтрализуют хлороводородной кислотой до удаления карбонатов:
Na2CO3 + 2HCl à 2NaCl + CO2 + H2O
Затем раствор, содержащий только натрия хлорид, упаривают до начала кристаллизации.
Источники получения калия хлорида — минералы сильвинит KCl × NaCl или карналлит KCl × MgCl2 × 6H2O. Из них выделяют калия хлорид методом флотации, а затем очищают, как и натрия хлорид. Идентификация. 1. Реакции на Cl- (а,b) 2. Реакции на Na+ (а,b,c) (для натрия хлорида)
2. Реакции на К+ (а,b,c) (для калия хлорида)
Количественное определение.
1. Прямая аргентометрия, точка эквивалентности устанавливается потенциметрически (ГФУ); пересчет на сухое вещество, (s=1): NaCl + AgNO3 à AgCl + NaNO3
2. Аргентометрия по Фольгарду в присутствии дибутилфталата (обратное титрование) (ГФУ); (s=1):
NaCl + AgNO3 à AgCl + NaNO3
AgNO3 + NH4SCN AgSCN + NH4NO3
3NH4SCN + Fe(NH4)(SO4)2 Fe(SCN)3↓ + 2(NH4)2SO4
3. Аргентометрия по Мору, прямое титрование, индикатор – калия хромат, (s=1):
NaCl + AgNO3 à AgCl + NaNO3
2AgNO3 + K2CrO4 à Ag2CrO4 + 2KNO3
3. Меркуриметрия; индикатор – дифенилкарбазон; (s=2): 2NaCl + Hg(NO3)2 àHgCl2 + 2NaNO3
Применение. Основная функция натрия хлорида – обеспечение постоянного осмотического давления крови. Калия хлорид используется при гипокалиемии (вследствие приема диуретиков); антиаритмическое средство. Хранение. В хорошо укупоренной таре.
7.Натрия гидрокарбонат. Получение, свойства, контроль качества, хранение и медицинское применение
Получают карбонаты и гидрокарбонаты аммиачно-хлоридным способом. На очищенный от примесей рассол хлорида натрия действуют аммиаком, а затем подвергают карбонизации в барботажных колоннах: NaCl + NH3 + CO2 + H2O à NaHCO3 + NH4Cl
Прокаливанием гидрокарбонатов получают карбонаты. Испытания на подлинность и количественное определение карбонатов и гидрокарбонатов основаны на химической реакции разложения минеральной кислотой: Na2CO3 + 2HCl à CO2 + 2NaCl + H2O
NaHCO3 + HCl à CO2 + NaCl + H2O
Этот процесс лежит в основе антацидного действия натрия гидрокарбоната.
Натрия гидрокарбонат был открыт в 1801 г. В. Розе. В настоящее время его получают при насыщении очищенного кристаллического карбоната натрия диоксидом углерода:
Na2CO3 · 10H2O + CO2 à 2NaHCO3 + 9H2O
Для окончательной очистки натрия гидрокарбонат перекристаллизовывают из теплой воды (60 °C), насыщенной диоксидом углерода. Натрия гидрокарбонат растворим в воде, практически нерастворим в этаноле. Водные растворы имеют слабощелочную реакцию.
Подлинность его устанавливают по наличию иона натрия и гидрокарбонат-иона. Последний обнаруживают с помощью реакции разложения разведенной хлороводородной кислотой, которое происходит с выделением пузырьков газа: NaHCO3 + HClàNaCl + H2O + CO2
Для количественного определения используют ту же реакцию. Навеску растворяют в свежее прокипяченной и охлажденной воде (воду кипятят для удаления растворенного углекислого газа) и титруют 0,1 М раствором хлороводородной кислоты (индикатор метиловый оранжевый). Натрия гидрокарбонат хранят в хорошо укупоренных банках.Натрия гидрокарбонат применяют в качестве антацидного средства внутрь, а также наружно в виде полосканий, промываний, ингаляций (0,5–2%-ные растворы).
8.Кальция хлорид и кальция сульфат гептагидрат. Получение, контроль качества, условия хранения.
Кальция хлорид получают обработкой мела или мрамора хлороводородной кислотой:
CaCO3 + 2HCl à CaCl2 + CO2 + H2O
В медицинской практике применяют кальция хлорид . Он очень легко растворим в воде с образованием растворов нейтральной реакции. Раствор в воде при этом сильно охлаждается. В отличие от многих неорганических солей кальция хлорид легко растворяется в этаноле.
Кальция хлорид-CaCl2 · 6H2O-Бесцветные кристаллы без запаха, горько-соленого вкуса, очень гигроскопичные, расплываются на воздухе, переходя при 34 °C в дигидрат. Наличие иона кальция устанавливают по окрашиванию бесцветного пламени горелки в кирпично-красный цвет и по образованию белого осадка при добавлении оксалата аммония к раствору кальция хлорида. Осадок растворим в разведенных минеральных кислотах, поэтому реакцию необходимо вести в нейтральной среде или в присутствии уксусной кислоты:

В разбавленных растворах ион кальция образует с серной кислотой (1:4) характерные игольчатые кристаллы CaSO4 × 2H2O.
Гексацианоферрат (II) калия при pH 7 в присутствии хлорида аммония образует с ионами кальция белый кристаллический осадок:
CaCl2 + K4[Fe(CN)6] + NH4Cl à KNH4Ca[Fe(CN)6]¯ + 3KCl
Кальция хлорид испытывают также на наличие хлорид-ионов.
Количественное определение кальция хлорида выполняют комплексонометрическим методом. В основе определения лежит тот же химический процесс, что и при анализе солей магния. Индикатором служит кислотный хром темно-синий, который в эквивалентной точке приобретает сине-фиолетовое окрашивание. Кальция хлорид можно количественно определить и по аниону аргентометрическим методом: CaCl2 + 2AgNO3 à 2AgCl¯ + Ca(NO3)2
При хранении необходимо учитывать высокую гигроскопичность кальция хлорида. Поэтому его хранят в небольших хорошо укупоренных стеклянных банках с пробками, залитыми парафином, в сухом месте. Кальция хлорид применяют в качестве средства, оказывающего противоаллергическое, противовоспалительное, кровоостанавливающее, диуретическое действие. В хирургической и стоматологической практике применяют кальция сульфат жженый (Calcii sulfas ustus). В природе широко распространен гипс, представляющий собой кальция сульфата дигидрат CaSO4·2H2O. При нагревании до 130–150 °C он теряет часть своей кристаллизационной воды и превращается в гемигидрат (полугидрат) 2CaSO4·H2O. Кальция сульфат жженый (гипс) — сухой, мелкий, аморфный порошок белого или слегка сероватого цвета. Он мало растворим (1:600) в воде, водный раствор имеет нейтральную реакцию. Подлинность гипса устанавливают по иону кальция и сульфат-иону. Гипс испытывают также на затвердевание: смесь 10 ч. гипса и 5 ч. воды должна затвердевать в белую твердую плотную массу не ранее, чем через 4 мин и не позднее чем через 10 мин. Хранят гипс в хорошо укупоренных стеклянных и жестяных банках в сухом прохладном месте.
9.Магния оксид и магния сульфат гептагидрат MgSO₄·7H₂O. Получение, контроль качества, условия хранения.
Встречается магний в природе в виде различных соединений, главным образом магнезита MgCO3, доломита MgCa(CO3)2, кизерита MgSO4 × H2O, эпсомита MgSO4 × 7H2O, различных силикатов (серпентин, асбест, тальк и др.). Соединения магния применяют в медицинской практике в виде магния оксида, магния сульфата и др.
Магния оксид можно получить при обработке природных рассолов гидроксидом кальция (известковым молоком). Образуется гидроксид магния, который превращают в оксид термической обработкой (при 500–700 °C):
MgCl2 + Ca(OH)2 à CaCl2 + Mg(OH)2¯
Mg(OH)2 à MgO + H2O
Приготовленный в таких условиях магния оксид называют «лёгкой магнезией». Магния сульфат получают нагреванием магнезита с избытком серной кислоты (избыток кислоты необходим, чтобы избежать образования основных солей магния): MgCO3 + H2SO4 à MgSO4 + H2O + CO2
При комнатной температуре из водных растворов кристаллизуется MgSO4 × 7H2O.
Магния оксид и магния сульфат различаются по физическим свойствам .Магния оксид-Белый мелкий легкий порошок без запаха. практически нерастворим в воде (свободной от примеси углекислого газа) и в этаноле, но растворим в разведённых кислотах. Магния сульфат -Бесцветные призматические выветривающиеся кристаллы .легко растворим в воде, практически нерастворим в этаноле.
Испытания на подлинность магния оксида проводят после предварительного растворения в разведенных кислотах: MgO + 2HCl à MgCl2 + H2O . Для обнаружения иона магния используют общую реакцию образования нерастворимого в воде, но растворимого в уксусной кислоте белого кристаллического осадка фосфата магния-аммония. Осадок выпадает при добавлении к раствору соли магния гидрофосфата динатрия и раствора аммиака:
NH4Cl
MgCl2 + Na2HPO4 + NH3 · H2О à NH4MgPO4¯ + 2NaCl + H2O
NH4MgPO4 + 2CH3COOH àMg(CH3COO)2 + NH4H2PO4
Ион магния в магния оксиде обнаруживают, осаждая его из растворов в хлороводородной кислоте избытком гидроксида натрия. Образующийся гидроксид магния представляет собой белый студенистый осадок, нерастворимый в избытке раствора гидроксида натрия.
В магния сульфате устанавливают наличие сульфат-иона: MgSO4 + BaCl2 à BaSO4¯ + MgCl2
 Соединения магния количественно определяют прямым
комплексонометрическим методом с использованием индикатора кислотного
хром черного специального (эриохром черный Т). После добавления индикатора
к титруемому раствору ионы магния образуют с ним непрочное комплексное
соединение:
Соединения магния количественно определяют прямым
комплексонометрическим методом с использованием индикатора кислотного
хром черного специального (эриохром черный Т). После добавления индикатора
к титруемому раствору ионы магния образуют с ним непрочное комплексное
соединение:
 Титрант — 0,05 М раствор трилона Б (ЭДТАNa2)
связывает находящиеся в растворе ионы магния в комплексное соединение:
Титрант — 0,05 М раствор трилона Б (ЭДТАNa2)
связывает находящиеся в растворе ионы магния в комплексное соединение:
Поскольку при этом происходит выделение серной кислоты, для поддержания оптимального значения pH среды необходимо прибавлять аммиачный буферный раствор. В эквивалентной точке, когда все ионы магния будут связаны в комплексное соединение металл — ЭДТА Na2, ЭДТА Na2 — металл, поэтому происходит разрушение комплекса индикатора с ионами магния. При этом красно-фиолетовая окраска раствора переходит в синюю окраску свободного индикатора:


Соединения магния хранят в хорошо укупоренной таре, так как магния оксид взаимодействует с углекислым газом и влагой, содержащимися в воздухе, образуя примесь карбоната и гидроксида магния: MgO + CO2 à MgCO3 MgO + H2O ® Mg(OH)2. Магния сульфат в плохо укупоренной таре постепенно теряет кристаллизационную воду.
10.Кислота борная и натрия тетраборат. Получение, контроль качества, условия хранения.
Наиболее важные минералы бора: борная кислота (сассолин) H3BO3; бура Na2B4O7 × 10H2O; кернит Na2B4O7 × 4H2O; борокальцит CaB4O7 × 4H2O; ашарит B2O3 × 2MgO × H2O и др. В медицинской практике применяют лекарственные вещества кислоту борную и натрия тетраборат. Источниками их получения являются природные минералы, которые либо сами содержат борную кислоту (сассолин) и натрия тетраборат (бура, кернит), либо разрушаются с их образованием.. Кислоту борную получают также разложением буры или борокальцита горячим раствором хлороводородной кислоты:
Na2B4O7 × 10H2O + 2HCl à4H3BO3 + 2NaCl + 5H2O
CaB4O7 × 4H2O + 2HCl + H2O à4H3BO3 + CaCl2
Фильтрат охлаждают, и выделившиеся кристаллы кислоты борной перекристаллизовывают из воды. Натрия тетраборат получают действием растворов карбоната натрия (при нагревании) на кислоту борную или минерал борокальцит:
4H3BO3 + Na2CO3 à Na2B4O7 + CO2 + 6H2O
CaB4O7 + Na2CO3àNa2B4O7 + CaCO3¯
При перекристаллизации натрия тетраборат присоединяет 10 молекул воды.
Кислота борная-H3BO3-Бесцветные, блестящие, жирные на ощупь чешуйки или мелкокристаллический порошок без запаха Натрия тетраборат (бура)Na2B4O7·10H2O представляют собой бесцветные , прозрачные вещества , легко выветривающиеся кристаллы или белый кристаллический порошок.
Водные растворы кислоты борной (1:50) имеют слабокислую реакцию (K = 6,4 × 10–10). В водной среде устанавливается равновесие: + H2O ⇄ B(OH)4– + H+
При нейтрализации гидроксидами щелочных металлов образуются соли тетраборной кислоты H2B4O7(тетрабораты) или реже метаборной HBO2 (метабораты).
Водные растворы натрия тетрабората имеют щелочную реакцию (вследствие гидролиза):
Na2B4O7 + 7H2O à 4H3BO3 + 2NaOH
Под действием сильных кислот происходит нейтрализация:
Na2B4O7 + 2HCl + 5H2O à 4H3BO3 + 2NaCl
Эту реакцию используют для подготовки натрия тетрабората к испытанию на подлинность, а также для его количественного определения. Подлинность соединений бора можно установить по реакции образования в присутствии этанола борноэтилового эфира:
 Для количественного определения соединений
бора используют кислотные свойства растворов кислоты борной в глицерине и
щелочные свойства водных растворов натрия тетрабората. При прямом титровании
кислоты борной щелочью образуется метаборат натрия, который в водных растворах
сильно гидролизуется: H3BO3 + NaOH à NaBO2 + 2H2O
Для количественного определения соединений
бора используют кислотные свойства растворов кислоты борной в глицерине и
щелочные свойства водных растворов натрия тетрабората. При прямом титровании
кислоты борной щелочью образуется метаборат натрия, который в водных растворах
сильно гидролизуется: H3BO3 + NaOH à NaBO2 + 2H2O
NaBO2 + 2H2O à H3BO3 + NaOH
Для количественного определения используют способность кислоты борной образовывать с глицерином сильную одноосновную диглицериноборную кислоту, которую можно с достаточной точностью оттитровать щелочью, используя в качестве индикатора фенолфталеин:
 Количественное определение кислоты борной проводят в
смеси (1:4) свежепрокипяченной воды
(свободной от углекислого газа) и нейтрализованного (по фенолфталеину)
глицерина при комнатной температуре. Для контроля полноты образования натриевой
соли диглицериноборной кислоты к концу титрования добавляют дополнительную
порцию (10 мл) глицерина. Сохранение при этом розовой окраски свидетельствует о
достижении эквивалентной точки.
Количественное определение кислоты борной проводят в
смеси (1:4) свежепрокипяченной воды
(свободной от углекислого газа) и нейтрализованного (по фенолфталеину)
глицерина при комнатной температуре. Для контроля полноты образования натриевой
соли диглицериноборной кислоты к концу титрования добавляют дополнительную
порцию (10 мл) глицерина. Сохранение при этом розовой окраски свидетельствует о
достижении эквивалентной точки.
Количественное определение натрия тетрабората выполняют методом нейтрализации (индикатор метиловый оранжевый), используя для этого реакцию взаимодействия с хлороводородной кислотой:
Na2B4O7 · 10H2O + 2HCl à 4H3BO3 + 2NaCl + 5H2O
Лекарственные препараты соединений бора хранят в защищённом от света месте, в хорошо укупоренной таре. Кислоту борную и натрия тетраборат применяют в качестве наружных антисептических средств в виде водных 1–4%-ных растворов.
11.Алюминия оксид гидратированный и алюминия фосфат. Получение, контроль качества, условия хранения.
Из соединений алюминия в медицине чаще всего применяют алюминия гидроксид. Источниками получения алюминия и его солей являются минералы боксит, гиббсит (гидраргиллит), алунит и другие, представляющие собой смесь гидроксидов или их сочетание с сульфатом алюминия, оксидом кремния (IV). Алюминия гидроксид получают из сульфата алюминия или квасцов и раствора аммиака:
Al2(SO4)3 + 6NH3 · H2О à 2Al(OH)3¯ + 3(NH4)2SO4
2KAl(SO4)2 + 6NH3 · H2О à 2Al(OH)3¯ + K2SO4 + 3(NH4)2SO4
Осаждают при температуре 60 °C в присутствии сульфата аммония. Осадок отфильтровывают, промывают горячей водой и высушивают при 40 °C. Известны четыре кристаллические (a, b, g и g’) модификации и аморфный (гелеобразный) алюминия гидроксид переменного состава Al2O3 · nH2O. По современным представлениям образование гидроксида алюминия происходит по схеме:
Al(H2O)63+ + H2OàAl(H2O)5OH2+ + H3O+ (pKa1 = 4,96)
2Al(H2O)4(OH)+2 + nH2O à Al2O3 × nH2O + 2H3O+
H3O+ + OH– à2H2O
Алюминиягидроксид Al(OH)3 или Al2O3 · nH2O -Аморфный рыхлый белый порошок . Алюминия гидроксид практически нерастворим в воде и этаноле, но растворим при нагревании в разведённых кислотах и растворах едких щелочей с образованием прозрачного или слабо мутного раствора:
Al(OH)3 + 3HCl à AlCl3 + 3H2O
Al(OH)3 + NaOH à Na[Al(OH)4]
Таким образом алюминия гидроксид является амфотерным соединением:
Al2O3 × nH2O + 6H+ à 2Al3+ + (3+n)H2O
Al2O3 × nH2O + 2OH– à Al(OH)4– + Al(OH)4(H2O)2+ + yH2O
Добавлением раствора хлорида аммония к щелочному раствору можно понизить pH раствора и осадить гидроксид алюминия: 2Al(OH)4– + NH4+ à Al2O3 · nH2O¯ + NH3. Эту реакцию используют для испытания на подлинность.. Ион алюминия обнаруживают с помощью реакции пиролиза. Смоченное раствором нитрата кобальта небольшое количество алюминия гидроксида образует после прокаливания плав, окрашенный в синий цвет вследствие образования алюмината кобальта (тенаровой сини) — Co(AlO2)2.
Количественное определение алюминия гидроксида выполняют комплексонометрическим методом, используя обратное титрование. Избыток ЭДТА Na оттитровывают 0,02 М раствором сульфата меди (индикатор мурексид). Для количественного определения алюминия гидроксида может быть применён гравиметрический метод, основанный на предварительном t°
прокаливании,охлаждении и взвешивании образовавшегося оксида алюминия: 2Al(OH)3 à Al2O3 + 3 H2O. Хранят алюминия гидроксид в хорошо укупоренной таре, применяют как антацидное средство. Применяют в медицине также алюминия фосфат (Aluminium phosphate). Его получают из растворимых солей алюминия при взаимодействии с эквивалентными количествами ортофосфатов щелочных металлов в растворах с pH 4,0-4,5: Al2(SO4)3 + 2Na3PO4 + nH2O à2AlPO4 · nH2O + 3Na2SO4 Выпадает аморфный осадок. Применяют алюминия фосфат в виде геля в качестве противоязвенного, антацидного, обволакивающего, адсорбирующего средства. Механизм действия заключается в том, что в желудке в течение 10 мин pH повышается до 3,0-5,0 и снижается протеолитическая активность пепсина. Назначают при различных заболеваниях желудочно-кишечного тракта внутрь, через 1-2 часа после еды по 3-5 пакетиков на приём (один пакетик с 16 г геля содержит 10,4 г алюминия фосфата). Хранят в сухом прохладном месте. Замораживание не допускается. Применяют как антацидное средство при язвенной болезни, гастритах, диспепсии и др.
12.Висмута нитрат основной. Получение, контроль качества, условия хранения.
Висмут в природе встречается в виде висмутина Bi2S3, бисмита Bi2O3, бисмутина Bi2CO3(OH)4, самородного висмута и др. Висмута нитрат основной получают окислением свободного от примесей металлического висмута концентрированной азотной кислотой: Bi + 4HNO3 à Bi(NO3)3 + 2H2O + NO
Нитрат висмута гидролизуется в кипящей воде с образованием нерастворимой соли висмута нитрата основного:

Висмута нитрат основной практически нерастворим в воде и этаноле. Однако смоченный водой, он окрашивает синюю лакмусовую бумагу в красный цвет вследствие гидролиза с образованием азотной кислоты и гидроксида висмутила:

Висмута нитрат основной растворим в кислотах (азотной, хлороводородной). При этом происходит процесс, обратный получению его из средней соли:

Подлинность висмута
нитрата основного устанавливают
прокаливанием, которое приводит к разложению с образованием желто-бурых паров
(диоксид азота) и желтого остатка (оксид висмут
При добавлении сульфида натрия к раствору висмута нитрата основного в минеральной кислоте выпадает коричнево-черный осадок (сульфид висмута): 2Bi(NO3)3+ 3Na2SàBi2S3¯ + 6NaNO3
Если взболтать около 0,1 г висмута нитрата основного с разведенной серной кислотой, а затем профильтровать, то после добавления к фильтрату 2 капель раствора иодида калия образуется черный осадок иодида висмута, растворимый в избытке реактива с образованием желто-оранжевого раствора комплексной соли: Bi2(SO4)3+ 6KIà 2BiI3¯ + 3K2SO4 ///// BiI3+ KIàKBiI4
Количественное определение выполняют комплексонометрическим методом. Навеску, растворенную в нагретой азотной кислоте, титруют 0,05 М раствором трилона Б (ЭДТА Na2) в присутствии индикатора ксиленолового оранжевого или пирокатехинового фиолетового.:

Выделяющаяся азотная кислота не мешает титрованию, так как соли висмута количественно взаимодействуют с ЭДТА Na2 при pH 2–4.
Хранят висмута нитрат основной в хорошо укупоренной таре, предохраняющей от действия света. При доступе влаги и света он постепенно гидролизуется с образованием азотной кислоты и оксидов азота. Висмута нитрат основной применяют как вяжущее и отчасти антисептическое средство при желудочно-кишечных заболеваниях (по 0,25–0,5 г).
13.Цинка оксид и цинка сульфат гептагидрат. Получение, контроль качества, условия хранения.
В медицине применяют неорганические соединения цинка: цинка оксид и цинка сульфат. Основной источник их получения — очищенный от примесей металлический цинк. Цинка сульфат впервые получен в 1730 г. растворением цинка в разведенной серной кислоте.: Zn + H2SO4àZnSO4 + H2
Из растворов кристаллизуется гептагидрат цинка сульфата при 39-41 °C. При 42-70 °C образуется гексагидрат, а выше 70 °C — моногидрат. Раствор цинка сульфата при нагревании с карбонатом натрия образует осадок основного карбоната цинка, который промывают для удаления сульфат-ионов, сушат и прокаливают (300 °C) до получения оксида:
5ZnSO4 + 5Na2CO3 + 3H2O à2ZnCO3 · 3Zn(OH)2¯ + 5Na2SO4 + 3CO2
300 °C
2ZnCO3 · 3Zn(OH)2 à5ZnO + 2CO2 + 3H2O
Цинка оксид получают также окислением цинка на воздухе или в кислороде:
2Zn + O2 à 2ZnO
Цинка окись, цинка оксид -ZnO- Белый или белый с желтоватым оттенком аморфный порошок Цинка сульфат- ZnSO4 · 7H2O- Бесцветные прозрачные кристаллы или мелкокристаллический порошок без запаха. На воздухе выветривается, а при 280 °C полностью теряет кристаллизационную воду. Соединения цинка проявляют амфотерные свойства. При растворении в разведенных минеральных кислотах цинка оксид образует соли, а в избытке растворов гидроксидов щелочных металлов растворимые в воде гидроксокомплексы:
2NaOH NaOH H2SO4
[Zn(OH)4]2– à Zn(OH)2 à ZnO à ZnSO4
Гидроксид цинка растворим также в избытке раствора аммиака с образованием комплексной соли:
Zn(OH)2 + 4NH4OH à [Zn(NH3)4](OH)2 + 4H2O
Перед испытанием на подлинность цинка оксид превращают в соль, растворяя в разведенной серной кислоте. Наличие иона цинка устанавливают по образованию белого осадка сульфида цинка, нерастворимого в уксусной кислоте и легко растворимого в разведенной хлороводородной кислоте (поэтому реакцию нужно выполнять в нейтральной среде): ZnSO4 + Na2S à ZnS¯ + Na2SO4
Эта реакция позволяет отличать цинк от других тяжелых металлов, образующих сульфиды черного цвета. Растворы солей цинка образуют белый гелеобразный осадок при взаимодействии с раствором гексацианоферрата (II) калия: ZnSO4+ K4[Fe(CN)6] àK2Zn[Fe(CN)6]¯ + K2SO4
Осадок нерастворим в разведенных кислотах, растворим в растворах щелочей. Тетрароданомеркурат (II) аммония образует с солями цинка в слабокислой среде белый кристаллический осадок: ZnCl2 + (NH4)2Hg(NCS)4 àZnHg(NCS)4¯ + 2NH4Cl
Подобно ионам алюминия (III) цинк во всех соединениях можно обнаружить специфичной реакцией, основанной на прокаливании с нитратом кобальта. Образуется так называемая «зелень Ринмана» — ярко-зелёный плав: t°
ZnO + Co(NO3)2àCoZnO2+ 2NO2 + ½O2
Количественное определение обоих лекарственных веществ проводят комплексонометрическим методом по иону цинка.. Соединения цинка хранят в хорошо укупоренной таре. При хранении следует иметь в виду, что цинка оксид поглощает углекислый газ из воздуха: ZnO + CO2 à ZnCO3
Растворы цинка сульфата при хранении мутнеют вследствие образования основной соли: 3Zn(OH)2 · ZnSO4 · 4H2O. Цинка оксид применяют наружно в качестве вяжущего, подсушивающего и дезинфицирующего средства при кожных заболеваниях. Растворы цинка сульфата (0,1–0,25%) применяют в качестве вяжущего и антисептического средства в офтальмологии, оториноларингологической и урологической практике.
14.Соединения серебра: серебра нитрат, протаргол. Получение, контроль качества, условия хранения.В медицинской практике применяют серебра нитрат и коллоидные препараты серебра: колларгол и протаргол. Получают серебра нитрат действием на металлическое серебро избытком азотной кислоты. Происходит процесс окисления серебра с образованием соли:
3Ag + 4HNO3à3AgNO3+ 2H2O + NO
Для очистки от примесей полученный серебра нитрат осаждают из раствора хлороводородной кислотой в виде хлорида серебра:
AgNO3 + HCl à AgCl¯ + HNO3
Хлорид серебра промывают и восстанавливают цинковой пылью до металлического серебра:
2AgCl + Zn + H2SO4à2Ag¯ + ZnSO4 + 2HCl
Затем вновь получают серебра нитрат из очищенного металлического серебра, растворяя его в азотной кислоте. Полученный раствор концентрируют до кристаллизации. Под влиянием света, особенно в присутствии следов органических веществ, серебра нитрат темнеет, так как происходит восстановление серебра: AgNO3 à Ag + NO + O2
Свойство - серебра нитрат-AgNO3-Бесцветные прозрачные кристаллы в виде пластинок или белых цилиндрических палочек, без запаха.
Для испытания подлинности серебра нитрата использованы те же принципы, что и при идентификации меди (II) сульфата: восстановление и способность к комплексообразованию. Серебро восстанавливается из аммиачного раствора серебра нитрата при нагревании с раствором формальдегида:
AgNO3 + NH3 · H2O à AgOH + NH4NO3
AgOH + 2NH3à [Ag(NH3)2]OH

Эти реакции лежат в основе образования «серебряного зеркала» — тонкой блестящей пленки серебра на стенках пробирки.
Ион серебра можно открыть с помощью реакции, которую используют для обнаружения хлорид-иона. Реактивом при этом служит раствор хлороводородной кислоты или хлорида натрия. Осаждающийся хлорид серебра нерастворим в азотной кислоте, но растворяется в растворе аммиака с образованием комплексного соединения:
AgNO3 + HCl ® AgCl¯ + HNO3
AgCl + 2NH3 · H2О à [Ag(NH3)2]Cl + 2H2O
Для обнаружения иона серебра можно применить реакцию осаждения с иодид-ионом. Образуется желтого цвета осадок иодида серебра, нерастворимый в растворах аммиака и азотной кислоты. Ион серебра можно обнаружить, используя реакцию осаждения с хроматом калия, по образованию оранжево-красного осадка хромата серебра: AgNO3 + K2CrO4 à AgCrO4¯ + 2KNO3
Количественно серебра нитрат определяют тиоцианатометрическим (роданометрическим) методом:
AgNO3 + NH4NCS à AgNCS¯ + NH4NO3
Избыток титранта — тиоцианата аммония взаимодействует с индикатором —железоаммониевыми квасцами, окрашивая смесь по окончании титрования в розовый цвет:
3NH4NCS + FeNH4(SO4)2 à Fe(NCS)3 + 2(NH4)2SO4
Серебра нитрат хранят по списку А в хорошо укупоренных банках с притертой пробкой, в защищенном от света месте, чтобы не допустить восстановления серебра нитрата до металлического серебра. Применяют серебра нитрат в качестве антисептического средства наружно в виде 1–2%-ных водных растворов для лечения глазных и кожных заболеваний. (Необходимо очень тщательно контролировать концентрацию.)
16. Меди сульфат пентагидрат. Получение, контроль качества, условия хранения.
Меди сульфат пентагидрат - CuSO4 • 5H2O Его можно получить, действуя серной кислотой на
металлическую медь в присутствии окислителей.. Металлическую медь растворяют в
нагретой разбавленной серной кислоте при продувании воздуха:
2Cu +2H2S04 + O2 —2CuS04 + 2Н20
В качестве окислителя можно использовать не кислород воздуха, а чистую азотную
кислоту:
3Cu + 3H2SO4 + 2HNO3 ----3CuSO4 + 4Н20 +2NO
Полученный раствор выпаривают досуха для удаления воды и оксидов
азота.
Для установления
подлинности препарата используют свойство меди легко восстанавливаться .В
качестве восстановителя берут железную пластинку или гвоздь, которые* при
соприкосновении с раствором меди (II) сульфата покрываются красным налетом
металлической меди:
Fe + CuSO4 —> Cu + FeSO4
Ион меди можно идентифицировать, используя способность этого элемента легко
образовывать комплексные соединения с раствором аммиака. Под действием аммиака
из раствора меди (II) сульфата вначале осаждается голубой осадок: 2CuSO4
+ 2NH3*Н2О —> Cu2(OH)2SO4I + (NH4)2SO4
Осадок легко растворяется в избытке раствора аммиака, образуя комплексное соединение
темно-синего цвета: Cu2(OH)2SO4 + (NH4)2SO4 + бNH4OH —> 2
[Cu(NH3)4] SO4 + SH2O
Препарат дает характерную реакцию на сульфат-ион.
Количественное определение препарата основано на восстановлении катиона
меди (II) до меди (I): 2CuS04 + 4KI —> 2CuI2l + 2K2S04
2CuI2 —> 2CuI + I2
2CuS04 + 4KI —> 2Cul +
I2 + 2 K2SO4
Содержание меди (II) сульфата в
препарате может быть также установлено комплексонометрическим методом. Препарат
хранят по списку Б в хорошо укупоренной таре, не допуская потери
кристаллизационной воды, что может привести к передозировкам при приготовлении
лекарственных форм.
15.Соединения железа: железа сульфат гептагидрат, железа хлорид гексагидрат. Получение, контроль качества, условия хранения
В медицинской практике применяют железа (II) сульфат. Гептагидрат сульфата железа FeSO4 · 7H2O содержит природный минерал мелантерит. Его получают также, растворяя избыток восстановленного железа в 25–30%-ном растворе серной кислоты, при нагревании до 80 °C: Fe + H2SO4 à FeSO4 + H2
Раствор упаривают до кристаллизации, и полученный железа (II) сульфат сушат при 30 °C.
Железа (II) сульфат проявляет восстановительные свойства, образует двойные соли (соль Мора), аддукты (FeSO4)x(NO)y, другие комплексы. С раствором аммиака или щелочей соли железа (II) образуют осадок железа (II) гидроксида светло-зеленого цвета, который на воздухе постепенно превращается в бурого цвета осадок железа (III) гидроксида:
Fe2+ + 2OH– à® Fe(OH)2¯
4Fe(OH)2 + O2 + 2H2O à 4Fe(OH)3
Свойства железа (II) сульфата FeSO4 · H2O--Прозрачные кристаллы светлого голубовато-зеленого цвета или кристаллический бледно-зеленый порошок. На воздухе выветривается. Катион железа (II) можно обнаружить с помощью различных реакций. ФС рекомендует для этого реакцию образования синего осадка турнбулевой сини при действии раствором гексацианоферрата (III) калия:
FeSO4 + K3[Fe(CN)6] à FeK[Fe(CN)6]¯ + K2SO4
C сульфид-ионами катионы железа (II) образуют черный осадок сульфида:
FeSO4 + Na2S àFeS¯ + Na2SO4
Диметилглиоксим образует с ионами железа (II) в аммиачных растворах устойчивое комплексное соединение красного цвета, растворимое в воде. Для количественного определения используют реакцию окисления ионов железа (II) в ионы железа (III) с помощью титрованного раствора перманганата калия: 10FeSO4+ 8H2SO4+ 2KMnO4à 5Fe2(SO4)3+ K2SO4+ 2MnSO4+ 8H2O
Использование хлороводородной кислоты вместо серной ведёт к перерасходу титранта, т.к. избыток хлоридов взаимодействует с перманганат-ионом. Простым методом, позволяющим быстро и точно определять содержание железа (II), является цериметрия. Соли железа (II) в присутствии разведенной серной кислоты и a,a’-дипиридила приобретают интенсивное красное окрашивание:

Ce4+ +e– àCe3+
2FeSO4 + 2Ce(SO4)2 à Fe2(SO4)3 + Ce2(SO4)3
Окраска исчезает после добавления избытка раствора сульфата церия (IV), что позволяет использовать a,a’-дипиридил в качестве индикатора при цериметрическом определении.
Фотометрический метод основан на образовании окрашенного комплекса железа (II) с о-фенантролином. Оптическую плотность измеряют при 508 нм. Титрование солей железа (II) может быть выполнено методом дихроматометрии по реакции:
6Fe2+ + Cr2O72– + 14H+ à 6Fe3+ + 2Cr3+ + 7H2O. В качестве индикатора используют дифениламин.
Железа (II) сульфат хранят в хорошо укупоренных банках в сухом месте, чтобы не допустить потери кристаллизационной воды. Он может также окисляться во влажном воздухе с образованием основной соли Fe2(OH)4SO4. При 64 °C железа (II) сульфат плавится в своей кристаллизационной воде.
ЛС средства органической природы
1.Глицерин. Получение, контроль качества, условия хранения.
Получение. Омыление жиров
 В присутствии щелочей или катализаторов образуются
глицерин и высокомолекулярные жирные кислоты.
Подлинность глицерина устанавливают по образованию непредельного
альдегида — акролеина под действием водоотнимающих веществ (Hапример гидросульфата
калия):
В присутствии щелочей или катализаторов образуются
глицерин и высокомолекулярные жирные кислоты.
Подлинность глицерина устанавливают по образованию непредельного
альдегида — акролеина под действием водоотнимающих веществ (Hапример гидросульфата
калия):

 Образование акролеина происходит также при нагревании
смеси глицерина с борной кислотой. Открывают глицерин с помощью реакции
образования глицерата меди» Смешивают предварительно 5% -ный раствор сульфата
меди с раствором гидроксида натрия. К выпавшему голубому осадку гидроксида меди
прибавляют несколько капель глицерина. Осадок растворяется с образованием
синего раствора глицерата меди.
Образование акролеина происходит также при нагревании
смеси глицерина с борной кислотой. Открывают глицерин с помощью реакции
образования глицерата меди» Смешивают предварительно 5% -ный раствор сульфата
меди с раствором гидроксида натрия. К выпавшему голубому осадку гидроксида меди
прибавляют несколько капель глицерина. Осадок растворяется с образованием
синего раствора глицерата меди.

Для количественного определения глицерина можно использовать реакцию образования сложного эфира:

Глицерин хранят в хорошо укупоренной таре, в прохладном месте» учитывая способность глицерина поглощать пары воды, содержащиеся в воздухе. Глицерин в виде 84—88%-ной смеси с водой при наружном применении оказывает смягчающее действие.
2.Диэтиловый эфир (эфир, эфир анестезирующий). Получение, контроль качества, условия хранения.
Современный промышленный синтез
диэтилового эфира проводят при нагревании до 135 0C смеси этилового спирта
и концентрированной серной кислоты в специальных аппаратах — эфиризаторах.
Процесс идет в несколько стадий. Вначале образуется этилсерная кислота
(этилсульфат):

Этилсерная кислота взаимодействует с избытком этилового спирта, образуя
диэтиловый эфир:
Полученный эфир отгоняют в
приемник.
Наличие пероксидов в эфире медицинском и эфире для наркоза устанавливают по реакции с иодидами:
H5C2–O–O–C2H5 + 2KI + H2O ¾® I2 + H5C2–O–C2H5 + 2KOH
 Подлинность лекарственных препаратов диэтилового эфира подтверждают по физическим константам: температуре
кипения и плотности. При испытании на чистоту в обоих лекарственных препаратах
устанавливают отсутствие или допустимые пределы примесей, образующихся при
производстве и хранении. Примесь кислот определяют нейтрализацией водного извлечения
Подлинность лекарственных препаратов диэтилового эфира подтверждают по физическим константам: температуре
кипения и плотности. При испытании на чистоту в обоих лекарственных препаратах
устанавливают отсутствие или допустимые пределы примесей, образующихся при
производстве и хранении. Примесь кислот определяют нейтрализацией водного извлечения
Примесь альдегидов определяют по реакции с реактивом Несслера:

+ 3KOH + K2HgI4 ¾®
Hg¯ + CH3COOK + 4KI + 2H2O

 У эфира медицинского не
допускается образования осадка. Может быть только помутнение нижнего слоя и
желто-бурая окраска раствора. У эфира для наркоза допускается лишь слабая
опалесценция, изменения окраски и помутнения реактива быть не должно. Оба
лекарственных препарата относятся к списку Б. Эфир медицинский хранят в хорошо
укупоренных склянках оранжевого стекла в защищенном от света месте, вдали от
огня. Эфир для наркоза хранят в условиях, исключающих воздействие кислорода
воздуха и образование пероксидных соединений, которые могут стать причиной его
самовоспламенения при комнатной температуре.
У эфира медицинского не
допускается образования осадка. Может быть только помутнение нижнего слоя и
желто-бурая окраска раствора. У эфира для наркоза допускается лишь слабая
опалесценция, изменения окраски и помутнения реактива быть не должно. Оба
лекарственных препарата относятся к списку Б. Эфир медицинский хранят в хорошо
укупоренных склянках оранжевого стекла в защищенном от света месте, вдали от
огня. Эфир для наркоза хранят в условиях, исключающих воздействие кислорода
воздуха и образование пероксидных соединений, которые могут стать причиной его
самовоспламенения при комнатной температуре.
3.Раствор формальдегида (формалин). Получение, контроль качества, условия хранения
В медицинской практике применяют раствор формальдегида (формалин) Формальдегид получают окислением метилового спирта кислородом воздуха, Смесь даров метилового спирта и воздуха пропускают через нагретые до 500—600°С трубки, наполненные катализатором (медь, серебро, кокс):
 После охлаждения формальдегид (бесцветный газ с острым
запахом) растворяют в воде до получения 36,5—37,5 % водного раствора, который
называют формалином,.
После охлаждения формальдегид (бесцветный газ с острым
запахом) растворяют в воде до получения 36,5—37,5 % водного раствора, который
называют формалином,.
Идентифицировать формальдегид можно с помощью реакций образования окрашенных продуктов взаимодействия с хромотроповой или салициловой кислотами в присутствии концентрированной серной кислоты. ГФ рекомендует для этого использовать салициловую кислоту (появляется красное окрашивание). Образующееся окрашенное соединение называется ауриновым красителем:

Формальдегид дает положительную реакцию с фуксинсернистой кислотой:::::.

Для установления подлинности раствора формальдегида ГФ рекомендует использовать общую на альдегиды реакцию восстановления серебра (реакцию серебряного зеркала»):



Общими для альдегидов являются и другие реакции окисления. Они дают положительную реакцию с реактивом Несслера, при нагревании происходит образование бурого осадка металлической ртути:

При взаимодействии с реактивом Фелинга (смесь водного раствора сульфата меди и щелочного раствора натрия-калия тартрата) после нагревания до кипения выпадает кирпично-красный осадок оксида меди (I):

Количественное определение формальдегида в растворе и хлоралгидрата можно провести, используя реакцию окисления альдегидов иодом в щелочной среде. Иод при этом образует гипоиодит (сильный окислитель):

Гипоиодит окисляет альдегиды до кислот:
Затем добавляют избыток серной кислоты, непрореагировавший гипоиодит превращается в иод, который оттитровывают тиосульфатом натрия:
 Аналогичный химический процесс происходит при окислении
формальдегида пероксидом водорода в щелочной среде: Эту реакцию используют
для определения формальдегида.
Аналогичный химический процесс происходит при окислении
формальдегида пероксидом водорода в щелочной среде: Эту реакцию используют
для определения формальдегида.
Окисл-е происх при цериметрическом опред-ии формальдегида • Дей-ют изб титр-го раствора сульфата церия (IV):
Избыток его оттитровывают раствором соли Мopa: (NH4)2SO4*FeSO4*6H2O
В условиях аптеки
формальдегид в растворах определяют рефрактометрическим методом.
Р-р формальдегида следует хранить в хорошо закрытых склянках при
температуре не ниже 4*9°С* При более низкой температуре происходит
полимеризация с образованием параформа [СН2О]п — твердого белого вещества. Для
предохранения от полимеризации к препарату добавляют до 1% метилового спирта .
4.Нитроглицерин. Получение. Контроль качества, условия хранения.
Общая формула этой группы
препаратов R-O-NO2. Получают эти
препараты используя реакцию этерификации. Исходными продуктами синтеза служат
соответствующие спирты, азотная кислота и концентрированная серная кислота
(дегидратирующее средство).
Нитроглицерин синтезируют при —15 °С, пропуская (тонкой струей)
безводный глицерин через смесь концентрированных серной и азотной кислот:
 Подлинность препаратов устанавливают по нитрат-ионам, которые образуются при
гидролизе. Используют в качестве реактива раствор дифениламина, который
нитратами окисляется до имониевой соли дифенилбензидина, имеющей голубую
окраску. Выделившийся глицерин нагревают с гидросульфатом калия.
Образуется акролеин, обладающий характерным острым запахом.
Подлинность препаратов устанавливают по нитрат-ионам, которые образуются при
гидролизе. Используют в качестве реактива раствор дифениламина, который
нитратами окисляется до имониевой соли дифенилбензидина, имеющей голубую
окраску. Выделившийся глицерин нагревают с гидросульфатом калия.
Образуется акролеин, обладающий характерным острым запахом.

Количественно определить содержание нитроглицерина можно с помощью реакции
омыления, которую выполняют в присутствии окислителя (пероксида
водорода): 
Методика фотометрического
определения нитроглицерина в лекарственных формах по ГФ основана на измерении
светопоглощения (при длине волны 410 нм) продукта взаимодействия
препарата с фенол-2,4-дисульфокислотой , Нитраты, взаимодействуя с фенол - 2,4
- дисульфокислотой в аммиачной среде, образуют окрашенную в желтый цвет
оpmo-хиноидную форму 6-нитрофенол“

Препараты нитроглицерина следует хранить вдали от огня. При получении, хранении нитроглицерина следует соблюдать большую осторожность, так как от удара или при нагревании до 180°С он взрывается вследствие образования большого количества газов:

Поэтому пролитый нитроглицерин нужно сразу же залить гидроксидом калия, происходит реакция омыления:

5.Моносахарид: глюкоза. Получение, контроль качества, условия хранения.
 Глюкоза находится в виноградном соке, в плодах и других
органах различных растений. Основным источникам получения глюкозы в
промышленности является крахмал, который гидролизуют в присутствии минеральных
кислот:
Глюкоза находится в виноградном соке, в плодах и других
органах различных растений. Основным источникам получения глюкозы в
промышленности является крахмал, который гидролизуют в присутствии минеральных
кислот:

Глюкозу можно также получить гидролизом сахарозы с участием спиртового раствора хлороводорода:

Глюкоза в выкристаллизовывается, а фруктоза остается в растворе. Молочный сахар получают из молочной сыворотки выпариванием с последующей перекристаллизацией из воды.
Подлинность:
1.ФС Реакция Фелинга при нагревании до кипения положительна.
2.ФС Т°пл. безводной глюкозы
3.ФС [α] удельное вращение 10% раствора глюкозы.
Особенности определения [α]: Оптическая активность растворов глюкозы некоторое время после приготовления раствора изменяется – устанавливается равновесие между таутомерными формами, т.е. идет мутаротация. Явление мутаротации объясняется тем, что при растворении глюкозы образуется ее альдегидная форма, через которую получаются α- и β- аномеры (эпимеры), имеющие различные углы вращения: 64% α-D(+)-глюкозы и 36% β- D(+)-глюкозы.

Нефармакопейные реакции: 1) в аптечной практике глюкоза при подщелачивании (без нагревания!!!) с раствором меди сульфата образует растворимый фиолетово-синий комплекс. При стоянии этого раствора происходит окислительно-восстановительная реакция с выделением кирпично-красного осадка Сu2O. Т.о. в этой реакции одновременно доказывается и спиртовая и альдегидная группы.
2) Конденсация с тимолом в среде конц. Н2SO4 → темно-красное окрашивание. При действии на глюкозу концентрированной серной или хлороводородной кислотой из нее образуется 5-оксиметилфурфурол, вступающий в реакцию конденсации с фенолами (резорцином, α-нафтолом) или ароматическими аминами, с образованием окрашенных продуктов:



Количественное определение 
1.Количественное определение глюкозы Содержание глюкозы определяют иодометрическим методом, основанном на окислении альдегидной группы щелочными растворами иода до образования натриевой соли глюконовой кислоты:


2.Один из титриметрических методов анализа моносахаридов основан на использовании реактива Фелинга.


 3.Поляриметрический метод определения сахаров основан на измерении угла вращения
поляризованного света. Угол вращения a (в градусах), измеряемый на поляриметре,
и удельное вращение
3.Поляриметрический метод определения сахаров основан на измерении угла вращения
поляризованного света. Угол вращения a (в градусах), измеряемый на поляриметре,
и удельное вращение  связаны
между собой уравнением:
связаны
между собой уравнением:  .
Зная удельное вращение, длину трубки l и измерив угол вращения,
можно вычислить массовую долю с (%) по формуле:
.
Зная удельное вращение, длину трубки l и измерив угол вращения,
можно вычислить массовую долю с (%) по формуле:
4.ГЖХ-метод определения глюкозы используют после превращения её в летучие соединения (ацетаты сорбита или нитрил глюконовой кислоты).
Хранение . в хорошо укупоренной таре при комнатной температуре. У субстанции (порошка) учитывают гигроскопичность, в водных растворах при хранении окисляется.
6.Кислота аскорбиновая. Получение, контроль качества, условия хранения.
Кислота аскорбиновая проявляет восстановительные свойства. В растворах под действием слабых окислителей различной природы она окисляется до дегидроаскорбиновой кислоты:
Процесс этот — обратимый. Кислотные свойства кислоте аскорбиновой придает наличие в молекуле двух енольных гидроксилов. Разрыва лактонного цикла в этих условиях не происходит, а образуются нейтральные растворимые монозамещенные соли
 Эта реакция лежит в основе определения кислоты
аскорбиновой методом нейтрализации.
Эта реакция лежит в основе определения кислоты
аскорбиновой методом нейтрализации.
Кислотные свойства кислоты аскорбиновой используют для испытания подлинности.
После добавления карбоната натрия в водном растворе происходит образование
ионизированной формы кислоты аскорбиновой. К полученной натриевой соли
прибавляют сульфат железа (II). Появляется темно-фиолетовое окрашивание,
обусловленное образованием аскорбината железа, Исчезающее после добавления
разведенной серной кислоты: -

При добавлении к ее раствору реактива Фелинга появляется оранжево-желтый осадок оксида меди (I). При взаимодействии препарата с раствором нитрата серебра его ион восстанавливается до металлического серебра (реакция образования «серебряного зеркала»):

Восстановительными свойствами кислоты аскорбиновой обусловлено превращение окрашенного в синий цвет раствора 2,6 -дихлорфенолиндофенола в бесцветное лейкооснование:

Количественно определяют кислоту аскорбиновую, используя в качестве титранта-окислителя 0,1 M раствор иодата калия в присутствии иодида калия:
Избыток иода окрашивает крахмал в синий цвет. Окисление кислоты аскорбиновой иодом до дегидроаскорбиновой кислоты лежит в основе иодометрического определения.:

В эквивалентной точке выделяется иод, окрашивающий крахмал в синий цвет:

Периметрическое определение основано на том же процессе окисления
кислоты аскорбиновой (в среде серной кислоты). Индикатором служит комплексное
соединение железа (II) с о-фенантролином. Оно имеет интенсивно-красное
окрашивание. В эквивалентной точке избыток сульфата церия
(IV) окисляет ион железа до трехзарядного и происходит образование
комплексного соединения голубого цвета:

Кислоту аскорбиновую хранят в хорошо укупоренной таре» предохраняя от действия света и кислорода воздуха.
7.Общая характеристика физико-химических и химико-аналитических и свойств лекарственных веществ, относящихся к аминокислотам и их производным. Кислота глутаминовая. Получение, контроль качества, условия хранения.
Общие физические и физ-хим свойства
Природные аминокислоты алифатического ряда – бесцветные, белые и желтоватого оттенка кристаллические субстанции, чаще хорошо растворимые в воде, Оптическая активность. Большинство аминокислот (кислота глутаминовая, цистеин, ацетилцистеин, пеницилламин) обладают оптической активностью, т.е. способностью вращать плоскость поляризованного луча света. Природные α-АК, входящие в состав белков, являются L-изомерами. Левовращающие аминокислоты обозначают L или (-), правовращающие D или (+). Не содержат асимметрического атома углерода и не обладают оптической активностью аминалон, пирацетам, кислота аминокапроновая. Метионин не обладает оптической активностью, т.к. является рацематом (смесью D и L – изомеров). Поглощение света в ИК- области спектра. Аминокислоты поглощают свет в ИК – области спектра (4000-400 см-1) и не обладают поглощением в УФ (нет хромофорных групп) – области спектра (220-350 нм).
 Общие химические свойства Кислотно-основные свойства. Аминокислоты обладают амфотерными свойствами. За счет
наличия аминогруппы (–NH2) c
неподеленной парой электронов
аминокислоты проявляют основные свойства, за счет подвижного атома
водорода карбоксильной группы (-COOH) –
кислотные свойства, поэтому
растворяются в кислотах и щелочах.
Общие химические свойства Кислотно-основные свойства. Аминокислоты обладают амфотерными свойствами. За счет
наличия аминогруппы (–NH2) c
неподеленной парой электронов
аминокислоты проявляют основные свойства, за счет подвижного атома
водорода карбоксильной группы (-COOH) –
кислотные свойства, поэтому
растворяются в кислотах и щелочах.
 1)Восстановительные свойства. Реакции
окисления. реакция окислительного дезаминирования с выделением аммиака и
диоксида углерода,
1)Восстановительные свойства. Реакции
окисления. реакция окислительного дезаминирования с выделением аммиака и
диоксида углерода,
Комплексообразующие свойства. За счет карбоксильной группы и аминогруппы аминокислоты способны вступать в реакции комплексообразования с солями тяжелых металлов в щелочной среде. Продукт взаимодействия АК с солями меди имеет темно-синюю окраску (образуется внутрикомплексная соль).

(водородные связи между медью и атомами азота)
ФП (ГФ ХII) Кислота глутаминовая Acidum glutaminicum (ЛН)Glutamic acid (МНН)
5 4 3 *2 1
HOOC – CH2 – CH2 - CH – COOH
│
NH2 L-2-Аминопентандиовая кислота (α- аминоглутаровая кислота)
Получение 1.Микробиологическим синтезом. Гидролиз белковых веществ и выделение АК хроматографическим методом 2. Синтетически, исходный продукт – аминослянный эфир:
Первый этап синтеза – ацетилирование (ацетиламиномалоновый эфир):

Второй – конденсация с акрилонитрилом

Третий – гидролиз

Четвертый – декарбоксилирование:

Подлинность.ФС ИК – спектр, снятый у субстанции ЛВ, должен соответствовать спектру стандартного об разца.ФС Удельное вращение [α] 5% раствора в разведенной НСI ( +30 - +34)
Реакции доказательства принадлежности ЛВ к группе аминокислот.ФС Нингидриновая реакция (проба). Продукты реакции большинства ЛВ окрашены в сине-фиолетовый цвет. Цистеин дает красное окрашивание. Первая стадия – разрушение глутаминовой кислоты, Дикетоксигидринден

Вторая
стадия – конденсация выделившегося
аммиака с нингидрином и его восстановленной формой
сине – фиолетовое окрашивание образуется аммонийная соль енольной формы дикетогидринденкетогидринамина (синь Роймана) и может применятся для идентификации некоторых ионов металлов – меди, кальция, серебра.
1.ГФ Х Реакция отличия глутаминовой кислоты от других представителей группы – сплавление глутаминовой кислоты с резорцином в присутствии конц серной кислоты. Смесь нагревают до появления зелено-коричневого окрашивания. Если теперь смесь охладить, добавить воды и раствора аммиака, то появится красно-фиолетовое окрашивание с зеленой флуоресценцией. При нагревании глутаминовая кислота переходит в пирролидонкарбоновую к-ту, которая конденсируется с резорцином:
 пиролидонкарбоновая
кислота
пиролидонкарбоновая
кислота

1Реакция комплексообразования с меди сульфатом (кислотные свойства). Образуется интенсивное синее окрашивание.

1.с растворами фенолфталеина, натрия гидроксида и формальдегида, добавляемых последовательно. Эффект реакции – появление, а при прибавлении формальдегида – исчезновение розового окрашивания.
Чистота
1. Прозрачность и цветность 10% раствора в в 1М НСI в сравнении с эталонными растворами
2. pH водного раствора (3,1-3,7). 3. Предельное содержание общих примесей - тяжелые металлы в сульфатной золе, мышьяк, хлориды. 4. Остаточные органические растворители. 5. Потеря в массе при высушивании не более 0,5% 6. Микробиологическая чистота, для нестерильных ЛС.
Специфические примеси 5. Недопустимые примеси посторонних АК определяют методом тонкослойной хроматографии. Проявляют нингидрином.
Количественного определения Алкалиметрический метод (вариант нейтрализации) определения аминокислот, содержащих две карбоксильные группы.
ФС ХII для кислоты глутаминовой (субстанция и таблетки). Алкалиметрия по бромтимоловому синему до перехода желтой окраски в голубовато-зеленую (рН 6,0 – 7,6) или нейтральному красному.
 Кстех
= 1/1; fэкв = 1,
Кстех
= 1/1; fэкв = 1,
Метод Кьельдаля 1 стадия. В колбу Кьельдаля помещают точную навеску вещества, добавляют концентрированную серную кислоту и смесь сульфата калия и сульфата меди (для увеличения Т°кипения), нагревают. Смесь кипятят до тех пор, пока раствор не просветлеет (станет светло-зеленым). и После этого продолжают сжигание (минерализацию) в течение 30 минут (для метионина 4-4,5 часа). Образуются неорганические соединения: вода, углекислый газ и сульфат аммония.

C→CO2; N→(NH4)2SO4→NH3
2 стадия. После этого к охлажденной смеси добавляют воду, подсоединяют колбу к парообразователю, добавляют избыток 30% гидроксида натрия (для нейтрализации H2SO4) и пропускают пар. Азот органического соединения, после минерализации его связанный серной кислотой в сульфат аммония, отгоняют с водяным паром количественно в виде аммиака в приемник с борной кислотой. Образуется смесь метабората и тетрабората аммония.
3 стадия. Смесь после полного окончания перегонки титруется 0,1М хлороводордной кислотой. Индикатор смешанный: метиловый красный и метиленовый синий 1:1 или м/о + м/с.
 Параллельно
проводят контрольный опыт.
Параллельно
проводят контрольный опыт.
3.Формольное титрование по Сёренсену (алкалиметрия после взаимодействия с формальдегидом). Для блокирования аминогруппы предварительно добавляют формальдегид, образуется устойчивое N-метиленпроизводное,. Индикатор – фенолфталеин или потенциометрически.

Кстех = 1/2; fэкв = 1/2; М.э. = М.м/2
1. Ацидиметрический метод неводного титрования В среде безводной уксусной кислоты подавляются кислотные свойства групп кислотного характера и усиливаются основные свойства групп основного характера.
2. Спектрофотометрический метод в видимой области спектра. Для получения окрашенных продуктов используют реакцию с нингидрином (гранулы кислоты глутаминовой для детей). Поляриметрический метод расчет по значению угла вращения и удельному вращению.
Хранение: ХУТ, защищающая от действия света. Применение Для лечения заболеваний ЦНС: эпилепсии, психозов, депрессивных состояний.
8.Глицин. Получение, контроль качества, условия хранения
Глицин (2-Аминоуксусная кислота) C2H5NO2. простейшая алифатическая аминокислота, единственная аминокислота, не имеющая оптических изомеров. Так же называется лекарственный препарат, состоящий из глицина и вспомогательных веществ (метилцеллюлоза водорастворимая, магния стеарат). Глицином («глицин-фото») также иногда называют параоксифениламиноуксусную кислоту, проявляющее вещество в фотографии. Белый или почти белый кристаллический порошок. Обладает полиморфизмом.Подлинность. 1.ИК-спектр. с калия бромидом, в области частот от 4000 до 400 см-1 2. Тонкослойная хроматография. Аминокислоты получают путем химического синтеза, биосинтеза или экстракцией из белковых гидролизатов. К химическому синтезу относят способ получения глицина через аммонолиз и последующее омыление водных растворов гликолонитрила Способ получения глицина щелочным гидролизом гидантоина Выход глицина составляет 95% , тем не менее ему присущи недостатки описанного способа, поскольку для получения исходного гидантоина необходима синильная кислота (синтез Штрекера), а его гидролиз требует количественных затрат водной щелочи. В промышленной практике наиболее распространен способ получения глицина аммонолизом монохлоруксусной кислоты (МХУК), , в водном растворе в присутствии гексаметилентетрамина
CI-CH2-COOH __ __> H2N-CH2-COOH+NH4Cl
__> H2N-CH2-COOH+NH4Cl
Количественное определение. Около 70 мг (точная навеска) субстанции растворяют в 3 мл муравьиной кислоты безводной, прибавляют 30 мл кислоты уксусной ледяной и немедленно после растворения титруют 0,1 М раствором хлорной кислоты. Конечную точку титрования определяют потенциометрически. 1 мл 0,1 М раствора хлорной кислоты соответствует 7,51 мг глицина C2H5NO2.
Хранение. В плотно закрытой упаковке, в защищенном от света месте при температуре не выше 25°С.
9.Кислота гамма-аминомасляная (аминалон). Получение, контроль качества, условия хранения
НФП Кислота гамма-аминомасляная Аминалон Gamma aminobutiric acid Aminalonum
4 (γ) 3(β) 2 (α) 1
H2N – CH2 - CH2 - CH2 – COOH
4 – Аминобутановая кислота (γ- аминомасляная кислота)
Получение Наиболее простой способ синтеза – гидролиз пирролидона-2 (ангидроформа ГАМК):

Подлинность
Физические и физико-химические показатели 1 ФС ИК – спектр должен соответствовать спектру, приведенному в ФС. 2.ФС Т°разложения.
Реакции
1.ФС реакция с нингидрином – синее (сине-фиолетовое) окрашивание.
Таблетки аминалона: реакция подлинности – нингидриновая проба после хроматографического отделения аминалона на пластинах Силуфол ТСХ.
2.ФС раствор препарата нейтрализуют по ф/фт раствором щелочи до розового окрашивания, затем добавляют формальдегид, также нейтрализованный до розового окрашивания по ф/фт, окраска исчезает

1.Реакция с роданидом калия (на аминалон специфическая). Смесь препарата и реактива нагревают, при этом фильтровальная бумага, импрегнированная ацетатом свинца над пробиркой, чернеет (сероводород). Реакция протекает по типу взаимодействия роданида калия с солями аммония:
KSCN + NH4+ → NH4SCN + K+
При нагревании роданид аммония перегруппировывается в изомерную тиомочевину:

Тиомочевина может переходить в имидную форму, разлагающуюся с выделением сероводорода:

H2S + Pb(CH3COO)2 → PbS↓ + 2CH3COOH
2.с сульфатом меди в щелочной среде – реакция образования окрашенных хелатных солей синего цвета.
Количественное определение 1.ФС Ацидиметрический метод неводного титрования для субстанции . Кислотно–основное титрование. Среда – ледяная уксусная к-та. Титрант – хлорная к-та, растворенная в ледяной уксусной к-те Инд. – кристаллический фиолетовый.
В среде безводной уксусной кислоты подавляются кислотные свойства групп кислотного характера и усиливаются основные свойства групп основного характера.
При растворении в протогенном растворителе - безводной уксусной кислотe цвиттер-ионная форма аминокислоты переходит в катионную. Растворение препарата:

Титрант:

Титрование и регенерация растворителя:


2. ФС
Таблетки аминалона 0,25 – формольное титрование, инд – ф/ф: 
Кстех = 1/1; fэкв = 1; М.э. = М.м
Хранение: ХУТ, сухое, защищенное от света место. Применение: ноотропное при ослаблении памяти, атеросклерозе мозговых сосудов, нарушении мозгового кровообращения.
10.Лекарственные вещества группы фенолов: фенол. Получение. Испытание на подлинность. Контроль качества. Порядок хранения.
Фенолы представляют собой производные ароматических углеводородов, которые содержат в молекуле одну или несколько гидроксильных групп, непосредственно связанных с ароматическим ядром. По числу гидроксильных групп различают одноатомные, двухатомные и трехатомные фенолы:


 Получение из каменноугольной кислоты : 1. Выщелачивание фенола из каменноугольной смолы щелочью.2.Вытеснение
фенола серной кислотой из фенолята натрия
Получение из каменноугольной кислоты : 1. Выщелачивание фенола из каменноугольной смолы щелочью.2.Вытеснение
фенола серной кислотой из фенолята натрия
 Определение подлинности фенолов базируется на реакциях, характерных для
фенольного гидроксила и для ароматического ядра. Фенол, в молекуле которого
содержится заместитель первого рода с электродонорными свойствами, бромируется
избытком бромной воды в орто- и пара- положения (электрофильное замещение).
Реакция протекает при комнатной температуре в отсутствие катализатора.
Определение подлинности фенолов базируется на реакциях, характерных для
фенольного гидроксила и для ароматического ядра. Фенол, в молекуле которого
содержится заместитель первого рода с электродонорными свойствами, бромируется
избытком бромной воды в орто- и пара- положения (электрофильное замещение).
Реакция протекает при комнатной температуре в отсутствие катализатора.
Взаимодействие с бромной водой


Индофенольная реакция. Появляется темно – синее окрашивание .

Взаимодействие с 4-аминоантипирином
Реакция протекает в присутствии щелочных окисляющих агентов

Реакция нитрозирования
Пп-нитрозофенол Бенбензохинон

Реагирует с избытком фенола образуется индофенольный краситель
Реакция нитрования с азотной кислотой

Растворы имеют желтую окраску, котрая усиливается в щелочной среде
Образование азокрасителей Реактивом является 0,1% -ный раствор сульфаниловой кислоты

Вначале реактив сульфаниловой кислоты обрабатывают нитритом натрия в солянокислой среде. Образуется соль диазония. Далее добавляют раствор фенола в аммиачной среде. Образуется азокраситель красного цвета . Количественное определение Реакции галогенирования применяют для количественного определения препаратов фенолов и их производных. Бромид-броматометрическоё определение фенола, тимола, резорцина выполняют обратным титрованием ОД M раствором бромата калия в присутствие бромида калия:
 Избыток титрованного раствора бромата калия приводит к
образованию эквивалентного количества брома. При определении фенола и резорцина
избыток брома устанавливают иодометрически:
Избыток титрованного раствора бромата калия приводит к
образованию эквивалентного количества брома. При определении фенола и резорцина
избыток брома устанавливают иодометрически:
Br2+ 2KI→I2+ 2KBr
I2+ 2Na2S2O→2NaI + Na2S4O6
Препараты фенолов можно также количественно определять иодометрическим и иодхлорометрическим методом. Сущность втих методов аналогична броматометрии:

Избыток иода титруют раствором тиосульфата натрия. Для количественного определения фенола и резорцина может быть использован цериметрический метод.
Препараты хранят в хорошо укупоренной таре. предохраняют от действия света,. Фенол хранят с предосторожностью по списку Б.
11.Резорцин. Получение, контроль качества, условия хранения.
Сульфированием бензола, например* получают резорцин:

Колич-е определ-е. Бромид-броматометрическоё определение фенола, тимола, резорцина выполняют обратным титрованием ОД M раствором бромата калия в присутствие бромида калия:


При определении резорцина избыток брома устанавливают иодометрически:

Для количественного определения резорцина может быть использован цериметрический метод.

Подлинность 1) При прибавлении к раствору препарата раствора хлорида окисного железа появляется сине-фиолетовое окрашивание, переходящее от прибавления раствора аммиака в буровато-желтое. 2) При сплавлении в фарфоровой чашке нескольких кристаллов препарата с избытком фталевого ангидрида получается плав желто-красного цвета. При растворении плава в растворе едкого натра появляется интенсивная зеленая флюоресценция. Препараты хранят в хорошо укупоренной таре. предохраняют от действия света. Фенол, резорцин и тимол применяют в качестве антисептических средств.
12. Производные п-аминофенола: парацетамол. Получение. Контроль качества, условия хранения. Парацетамол представляет собой белое кристаллическое вещество, умеренно растворимое в воде, легко растворимое в этаноле, растворимое в ацетоне и растворах едких щелочей, практически нерастворимое в эфире. Синтез парацетамола выполняют ацетилированием n-аминофенола:

Известен также способ синтеза парацетамола из фенола:

Подлинность парацетамола 1.ИК-спектру (снятому в вазелиновом масле) в области 4200-400 см–1, полосы поглощения которого должны полностью совпадать с прилагаемым к ФС рисунком спектра. 2.УФ-спектр 0,0005%-ного раствора парацетамола в метаноле, подкисленном хлороводородной кислотой, в области 220-350 нм имеет максимум поглощения при длине волны 249 нм. Водный раствор имеет максимум поглощения при 243 нм, а раствор в 0,001 М гидроксиде натрия — два максимума при 257 и 273 нм.
Цветную реакцию на фенольный гидроксил с раствором хлорида железа (III) используют для испытания подлинности парацетамола, в присутствии которого возникает сине-фиолетовое окрашивание. Испытание основано на реакциях гидролиза, окисления и образования производных индофенола:

Непрореагировавший n-аминофенол при взаимодействии с хинонимином образует индофенол:

Раствор парацетамола при действии азотной кислотой приобретает желто-бурую окраску.
Под действием реактива
Марки парацетамол приобретает
буро-красное окрашивание. Образовавшаяся в результате гидролиза парацетамола
соль ароматического амина после добавления нитрита натрия и щелочного раствора
b-нафтола образует красного цвета азосоединение за счет наличия в молекуле ароматической
аминогруппы Парацетамол также
образует красного цвета азосоединение с диазореактивом за счет наличия в
молекуле фенольного гидроксила:
Парацетамол также
образует красного цвета азосоединение с диазореактивом за счет наличия в
молекуле фенольного гидроксила:
 Парацетамол при кипячении (2 мин) с разведенной
хлороводородной или серной кислотой вследствие гидролиза выделяет уксусную
кислоту, которую можно обнаружить по запаху:
Парацетамол при кипячении (2 мин) с разведенной
хлороводородной или серной кислотой вследствие гидролиза выделяет уксусную
кислоту, которую можно обнаружить по запаху:

Реакцию кислотного гидролиза используют для испытания на подлинность и в различных способах количественного определения. Парацетамол количественно определяют по образующемуся при кипячении с обратным холодильником в течение 1 часа продукту кислотного гидролиза — гидрохлориду п-аминофенола, используя нитритометрический метод:

Эквивалентную точку устанавливают потенциометрически или с помощью внешнего индикатора — иодкрахмальной бумаги (ФС), которая синеет от выделившегося при добавлении избытка титранта иода:
NaNO2 + HCl à NaCl + HNO2 2)KI + HCl ----> KCl + HI 3)2HNO2 + 2HI àI2 + 2NO + 2H2O
Эквивалентную точку при нитритометрическом определении парацетамола можно также установить со смешанным внутренним индикатором, содержащим 0,1%-ный раствор тропеолина 00 и 0,15%-ный раствор метиленового синего.
Способ обратного цериметрического определения парацетамола основан на предварительном кислотном гидролизе и последующем окислении п-аминофенола избытком 0,1 М раствора сульфата церия. Процесс идет по схеме:

Точку эквивалентности устанавливают иодометрическим методом, добавляя 10%-ный раствор иодида калия и титруя выделившийся иод 0,1 М раствором тиосульфата натрия (индикатор крахмал): 2Ce(SO4)2+ 2KIàI2+ Ce2(SO4)3+ K2SO4. Фотоколориметрический метод определения парацетамола основан на образовании молибденовой сини при взаимодействии с молибдатом аммония в сильнокислой среде. Парацетамол хранят по списку Б в хорошо укупоренной таре, в сухом месте. Предохраняют от действия света, чтобы не допустить гидролиза и окисления.
13.Кислота бензойная и бензоат натрия. Получение. контроль качества, условия хранения.
Кислоты бензойную получают, используя общие методы синтеза ароматических кислот. Синтезируют кислоту бензойную, окисляя толуол различными окислителями — азотной или хромовой кислотами, дихроматом калия, диоксидом марганца:

Современный промышленный способ основан на жидкофазном окислении толуола кислородом воздуха при 130-160 °C и давлении 308-790 кПа по схеме:

 Натрия бензоат получают, выпаривая досуха раствор
соответствующей кислоты (бензойной или салициловой), нейтрализованной
эквивалентным количеством карбоната или гидрокарбоната натрия:
Натрия бензоат получают, выпаривая досуха раствор
соответствующей кислоты (бензойной или салициловой), нейтрализованной
эквивалентным количеством карбоната или гидрокарбоната натрия:

 Полученную соль перекристаллизовывают из спирта.
Полученную соль перекристаллизовывают из спирта.
Подлинность УФ-спектр водного раствора натрия бензоата в области 220-300 нм должен иметь максимум поглощения при 226 нм. Раствор (0,001%) кислоты салициловой в 0,5 М растворе серной кислоты имеет два максимума поглощения при 235 и 300 нм.
Кислота бензойная и натрия бензоат дают характерную реакцию с раствором хлорида железа (III). Кислоту бензойную предварительно растворяют в 0,1 М растворе гидроксида натрия (реакция раствора должна быть нейтральной). В результате реакции образуется нерастворимый в воде основной бензоат железа (III) розово-желтого цвета:

Подлинность натрия бензоата устанавливают по иону натрия (окраска бесцветного пламени горелки в желтый цвет) и по выделению соответствующих кислот после нейтрализации растворов натриевых солей разведенной азотной кислотой. Способы количественного определения бензойной основаны на использовании алкалиметрического метода. В качестве растворителя используют этанол (так как кислоты мало растворимы в воде). Этанол предварительно нейтрализуют по фенолфталеину (феноловому красному). Затем растворяют навеску и титруют 0,1 М раствором гидроксида натрия (не содержащим карбонатов) с тем же индикатором:

Натрия бензоат, количественно определяют ацидиметрическим методом. Титруют раствором хлороводородной кислоты, используя смешанный индикатор (смесь равных количеств метилового оранжевого и метиленового синего:

Хранят ароматические кислоты и их соли в сухом, защищённом от света месте, при комнатной температуре, в хорошо укупоренной таре, учитывая возможность возгонки кислоты бензойной и кислоты салициловой. Применяют кислоты бензойную и салициловую наружно в качестве антисептических средств. Натрия бензоат назначают как отхаркивающее средство (в микстурах по 0,2–0,5 г или внутривенно в виде 15%-ного раствора).
14. Кислота салициловая и салицилат натрия. Получение, контроль качества, условия хранения. Кислоты салициловую получают, используя общие методы синтеза ароматических кислот.
 В химической промышленности кислоту салициловую
получают карбоксилированием фенола по реакции Кольбе–Шмидта:
В химической промышленности кислоту салициловую
получают карбоксилированием фенола по реакции Кольбе–Шмидта:
При карбонизации фенолята калия происходит образование смеси салициловой и n-оксибензойной кислот. При использовании фенолята натрия получается в основном кислота салициловая. При синтезе кислоты салициловой могут образовываться также небольшие количества оксидифенила:
Натрия салицилат получают, выпаривая досуха раствор соответствующей кислоты (бензойной или салициловой), нейтрализованной эквивалентным количеством карбоната или гидрокарбоната натрия:
Полученную соль перекристаллизовывают из спирта.
Подлинность натрия салицилата подтверждают с помощью ИК-спектра в области 4000-400 см–1 (спресованный в таблетках с бромидом калия), который должен полностью совпадать со спектром, прилагаемым к ФС. Раствор (0,001%) кислоты салициловой в 0,5 М растворе серной кислоты имеет два максимума поглощения при 235 и 300 нм. Для испытания подлинности кислоты салициловой и натрия салицилата также используют в качестве реактива раствор хлорида железа (III):

С раствором сульфата меди натрия салицилат образует зеленого цвета салицилат меди:

При нагревании кислоты салициловой или натрия салицилата с концентрированной серной кислотой и метанолом возникает резкий характерный запах метилсалицилата. При нагревании выше температуры плавления кристаллов кислоты салициловой или нагревании ее смеси с кристаллами солей органических кислот (цитрата или ацетата натрия) происходит разложение с образованием фенола (запах) и диоксида углерода:

Диоксид углерода можно обнаружить по образованию опалесценции при пропускании выделяющегося газа через известковую воду: Ca(OH)2 + CO2--à CaCO3¯ + H2O
Кислота салициловая образует окрашенное в красный цвет соединение (ауриновый краситель) при действии раствором формальдегида в присутствии концентрированной серной кислоты.
Подлинность натрия салицилата устанавливают по иону натрия (окраска бесцветного пламени горелки в желтый цвет) и по выделению соответствующих кислот после нейтрализации растворов натриевых солей разведенной азотной кислотой.
Способы количественного определения салициловой кислот основаны на использовании алкалиметрического метода. В качестве растворителя используют этанол (так как кислоты мало растворимы в воде). Этанол предварительно нейтрализуют по фенолфталеину (феноловому красному). Затем растворяют навеску и титруют 0,1 М раствором гидроксида натрия (не содержащим карбонатов) с тем же индикатором:

Натрия салицилат количественно определяют ацидиметрическим методом
.

Количественное определение кислоты салициловой и натрия салицилата можно выполнить также бромид-броматометрическим методом, так как они являются производными фенола: KBrO3 + 5KBr + 3H2SO4--à 3Br2 + 3K2SO4 + 3H2O
 Избыток брома затем определяют иодометрическим
методом:
Избыток брома затем определяют иодометрическим
методом:
Br2 + 2KI --à I2 + 2KBr
I2 + 2Na2S2O3 --à 2NaI + Na2S4O6
Определить содержание кислоты салициловой и натрия салицилата можно иодхлорометрическим методом, основанным на иодировании этих веществ в орто- и пара-положениях до дииодпроизводных:

Затем прибавляют 10%-ный раствор иодида калия, который взаимодействует с избытком иодмонохлорида и выделившийся иод оттитровывают 0,1 М раствором тиосульфата натрия: 1)ICl + KI --à I2 + KCl 2)I2 + 2Na2S2O3--à2NaI + Na2S4O6
Хранение: в сухом, защищ от света месте, при комнатной т, в хорошо укупоренной таре.. Кислоту салициловую назначают в качестве антисептического и кератолитического средства наружно. Натрия салицилат оказывает противоревматическое, противовоспалительное, болеутоляющее и жаропонижающее действие при приеме внутрь в дозах по 0,5–1,0 г.
15.Натрия пара-аминосалицилат. Получение, контроль качества, условия хранения
ФП Натрия пара-аминосалицилат (ПАСК-Nа) Natrii рага- aminosalicylas
Натриевая соль п-аминосалициловой кислоты

ФГ в структуре вещества: NН2 - первичная ароматическая аминогруппа, ОН - фенольный гидроксил, СООН - карбоксильная группа
Получение – синтез, источник – м-аминофенол.
Процесс синтеза складывается из трех стадий: 1.Карбоксилирование м-аминофенола. 2.Выделение пара-аминосалициловой кислоты. 3.Получение натрия п-аминосалицилата. На первой стадии происходит карбоксилирование м-аминофенола по методу Кольбе-Шмидта (получение салициловой кислоты). На второй стадии под действием 50% серной кислоты происходит выделение ПАСК. На третьей стадииПАСК нейтрализуется гидрокарбонатом натрия и образуется ПАС-натрия.

Источником синтеза может быть нитробензол:

Описание: белый или белый с желтоватым оттенком или розоватый мелкокристаллический порошок. Легко растворим в воде, растворы при стоянии темнеют. Трудно растворим в спирте.
Подлиннность
1.УФ-спектрофотометрия. УФ-спектр 0,001% водного раствора ПАСК-Nа, который имеет 2 максимума поглощения– 265нм и 299нм (свидетельствует о наличии фенольного гидроксила и ПАА). ФС Соотношение оптических плотностей при длинах волн 265 и 299 нм должно быть в пределах 1,50-1,56 (указывает на отсутствие поглощающих при данных длинах волн примесей). Величины удельных показателей поглощения при максимумах поглощения равны соответственно 736 и 483.
2. Температура плавления Т°пл=122оС.
Реакции подлинности
1. ФС Реакция на фенольный гидроксил – в кислой средес раствором хлорида железа (III): образуетсякомплексное соединение (фиолетово-красное окрашивание).

Водные растворы ПАС-натрия имеют рН 6,5-8,5, в этой среде с хлоридом железа образуется комплекс коричнево-красного цвета. Поэтому проводят подкисление разведенной хлороводородной кислотой. 2.ФС Реакция образования азокрасителя, основанная на наличии в структуре: первичной ароматической аминогруппы и фенольного гидроксила. А)В методике ФС после диазотирования раствором нитрита натрия в кислой среде проводят азосочетание с β-нафтолом в щелочной среде, появляется красное окрашивание (ЛВ диазосоставляющая):

Б) в методике МФ-IIпосле диазотирования ЛВ в качестве азосоставляющей берется α-нафтиламин. Реакция проводится в кислой среде. Образуется красное окрашивание, переходящее в оранжевое при добавлении раствора гидроксида натрия (ЛВ диазосоставляющая):

В) следующий вариант образования азокрасителя основан на наличии фенольного гидроксила.
Г) есть еще один вариант, основанный на том, что ПАС-натрия имеет в структуре одновременно ПАА и фенольный гидроксил. В щелочной среде сочетается как фенол:
 В кислой среде ПАС-натрия сочетается как амин,
присоединение идет в 3-положении (пара- к ПАА).
В кислой среде ПАС-натрия сочетается как амин,
присоединение идет в 3-положении (пара- к ПАА).
3.На ион натрия–окрашивает пламя горелки в желтый цветФС.
 4. Реакция на карбоксильную группу – выделение свободной ПАСК(слабая к-та) из ее соли
минеральной кислотой:
4. Реакция на карбоксильную группу – выделение свободной ПАСК(слабая к-та) из ее соли
минеральной кислотой:
5. Реакция на карбоксильную и гидроксильную группы – с солями тяжелых металлов (с меди (II) сульфатом образует травянисто-зелёное окрашивание
Чистота 1.Прозрачность и цветность раствора, т.к. ПАС-натрия – лабильное вещество.
2.м-аминофенол (допустимая в пределах эталона примесь). Источников появления этой примеси два – 1) процесс получения (исходное вещество синтеза) и 2) процесс декарбоксилирования, который идет уже при 70-80оС.

Количественное определение
1.Фармакопейный метод – нитритометрический, основанный на реакции диазотирования первичной ароматической аминогруппы натрия нитритом в кислой среде в присутствии катализатора - калия бромида. индикатор внешний – йодкрахмальная бумага. Титруют до появления синего пятна на бумаге.
 йодкрахмальная бумага
йодкрахмальная бумага
NaNO2 + HCl → HNO2 + NaCl
KI + HCl → HI + KCl
2HNO2 + 2HI → I2+ 2NO↑ + 2H2O
Суммарно:
2NaNO2 + 2KI + 4HCl → I2 + 2NO↑ + 2NaCl + 2KCl + 2H2O
1.Броматометрический и йодхлорметрический методы (см. определение производных п-аминобензойной кислоты). f = ¼ 2.Ацидиметрия (см. определение салицилата натрия).f = 1
Хранение Хранят в хорошо укупоренной таре, в сухом, защищенном от света месте, предохраняя от окисления и образования примесей продуктов разложения. Применение Внутрь для лечения различных форм туберкулёза по 2,0-3,0 г.
18.Производные фенилпропионовой кислоты: ибупрофен. Синтез. Физические свойства. Определение подлинности. Испытание на чистоту. Количественное определение. Условия хранения. Фармакологическое действие.
d,l-2-(4-изобутилфенил)-пропионовая кислота

Описание белый или почти белый кристаллический порошок с характерным запахом. Тпл=75-77,5оС. Практически не растворим в воде, растворим в органических растворителях.
Подлинность
1.ИК-спектр ФС, снятый в диске с калия бромидом, в области 4000-400 см-1 должен иметь полное совпадение полос поглощения с полосами поглощения спектра стандартного образца.
2.УФ-спектр ФС раствора препарата в 0,1М натрия гидроксиде имеет максимумы при длинах волн 265 и 273 нм, а также плечо от 257 до 261 нм.
3.ВЭЖХ метод ФС.
4.ТСХ в таблетках – сравнение со стандартом (свидетелем).
Чистота 1. Цветность определяют спектрофотометрическим методом.
Оптическая плотность указанного раствора при длине волны λ=440 нм должна быть не более 0,28.
2.Посторонние примеси определяют: методом ГЖХ (по отношению суммы площадей пиков, обусловленных примесями, к площади пика ибупрофена). методом ТСХ
Количественное определение
1.ФС Алкалиметрия, вариант нейтрализации (по карбоксильной группе), растворитель и среда – этанол, индикатор – – тимоловый синий (от желтого до голубовато-серого).
 R-СООН + NаОН → R-СООNа+Н2О f
экв,ибупрофена =1
R-СООН + NаОН → R-СООNа+Н2О f
экв,ибупрофена =1
Ставят контрольный опыт (титруют без ЛВ)
2.ФС Метод ВЭЖХ (таблетки, растворы для инъекций). Количественное содержание рассчитывают по площади пика, которая пропорциональна количеству ЛВ в пробе.
3.СФМ в УФ-области (мази, таблетки).
Хранение Список Б, в сухом, защищенном, от света месте при комнатной температуре, в хорошо укупоренной ,таре Применение Противовоспалительное, анальгезирующее, жаропонижающеедействие. Применяют при ревматоидном артрите, остром ревматизме, артрозах (заболеваниях суставов) и невралгиях.

17. Производные фенилуксусной кислоты: диклофенак натрия. Синтез (уравнения хим. реакций). Физические свойства. Определение подлинности: физические и химические методы. Испытание на чистоту. Количественное определение. Условия хранения. Фармакологическое действие.
Diclofenac – Natrium (Ortophenum, Voltaren) 2-[(2,6-дихлорфенил)-амино]-фенилуксусной кислоты натриевая соль

Получение – синтез
1)исходные вещества – 2,6-дихлорацетанилид и бромбензол

2)конденсацией 2-хлорфенилуксусной кислоты с 2,6-дихлоранилином с последующей нейтрализацией образующейся кислоты рассчитанным количеством натрия гидроксида:

Описание
Белый или с кремоватым оттенком до светло-кремового цвета кристаллический порошок без запаха.
Подлинность
1. ИК -спектр, снятый в диске с калия бромидом, в области 4000-400 см-1 должен иметь полное совпадение полос поглощения с полосами поглощения спектра стандартного образца.
2. УФ-спектр в области 240-350 нм в растворе 0,1М натрия гидроксида имеет максимум поглощения при 276±2 нм и минимум при 249±2 нм
Реакции подлинности
1Реакция вытеснения органической кислоты сильной минеральной кислотой из ее соли и образование индолинона.

2Ион натрия доказывают по окрашиванию бесцветного пламени в желтый цвет.
3Ионы хлорав фильтрате после восстановительной минерализации ковалентно связанного хлора (прокаливания в тигле и растворения остатка в воде) дают реакцию с раствором серебра нитрата.
4Реакции окисления: а) Реактив Марки при наслаивании на раствор препарата образует зелено-белое кольцо б) При действии окислителей С нитритом натрия (на фрагмент дифениламина) – синее окраш-е .в) При обработке кристаллов конц серной кислотой на часовом стекле, они приобретают малиновое окрашивание.
5Реакции соле- и комплексообразования (по карбоксильной и амино- группе).
С нитратом серебра АgNО3 – белый осадок
С хлоридом железа (III) FеСl3 – жёлто-коричневый осадок
С сульфатом меди CuSО4 – светло-зеленый осадок
Чистота
1. Цветность определяют спектрофотометрическим методом. Оптическая плотность указанного раствора при длине волны λ=440 нм должна быть не более 0,28.
2. Специфические примеси (продукты синтеза – наличие соответствующей кислоты и индолинона) определяют методом ВЭЖХ.
Подвижная фаза: метанол – 0,1% раствор ортофосфорной кислоты (6:4). Детектор – спектрофотометр (длина волны 254 нм).
Количественное определение
1.Ацидuметрический мeтод титрования в среде протогенного растворителя (неводное титрование). Титруют хлорной кислотой в среде ледяной уксусной кислоты (субстанция диклофенака-натрия). Точку эквивалентности устанавливают по ФС потенциометрически или по индикатору кристаллическому фиолетовому. f .экв. (диклофенана-натрия) = 1. Суммарно:

Титрант: 
2. Спектрофотометрия в УФ-области спектра (лекарственные препараты: мази, таблетки).
3. Метод ВЭЖХ для диклофенака-натрия (таблетки, растворы для инъекций).
4.Ацuдuметрическuй метод титрования в водной среде (вариант вытеснения) в присутствии эфира, который извлекает органическую кислоту и тем самым сдвигает равновесие реакции вправо
R–СООNа + НСl → R–COOH↓ + NaCl f экв. = 1
Хранение в сухом, защищенном, от света месте при комнатной температуре, в хорошо укупоренной, таре. Применение противовоспалительное, жаропонижающее, анальгезирующее. Раствор в ампулах, таблетки, мази и гели для наружного применения.
16.Тетракаин гидрохлорид. Химическая формула. Названия. Физические свойства тетракаина гидрохлорида. Определение подлинности препарата: физические и химические методы (уравнения реакций, аналитические эффекты). Количественное определение препарата. Условия хранения. Фармакологическое действие.
1.Тетракаина гидрохлорид (ФП Дикаин)
Dicainum (ЛН)Tetracainehydrochloride (МНН)

2- Диметиламиноэтилового эфира 4-бутиламинобензойной кислоты гидрохлорид
β-диметиламиноэтилового эфира п-бутиламинобензойной кислоты гидрохлорид
Получение Синтез
1. получение п-бутиламинобензойной кислоты :

2. получение хлорангидрида п-бутиламинобензойной кислоты:

3. конденсация с диметиламиноэтанолом:

Описание: белый кристаллический порошок без запаха. Легко растворим в воде и спирте, трудно растворим в хлороформе, практически не растворим в эфире.
Подлинность
1. ФСУФ-спектр поглощения в области 220-350нм в присутствий фосфатного буфера с рН6 имеет макс поглощения при 227 нм и мин поглощения при 249 нм.
2. ФСИК-спектр в области 4000 – 400 см -1 в калия бромиде.
3. ФС и ГФХ Т°пл 147-150 градусов в цельсий
В структуре дикаина имеется вторичная ароматическая аминогруппа, сложно-эфирная группа и третичная алифатическая аминогруппа.
1. Реакции на вторичную ароматическую аминогруппу:
реакция образования азокрасителя отрицательная. Дикаин содержит вторичную ароматическую аминогруппу:
1.ГФХреакция Витали-Морена дикаина смачивают конц. азотной кислотой образуется нитропроизводное дикаина желтого цвета. Затем охлажденный остаток обрабатывают спиртовым раствором гидроксида калия, образуется соль аци-формы, имеющая хиноидную структуру (калиевая соль орто-хиноидного соединения) и окрашенная в кроваво-красный цвет:


2.ГФХкак соль азотистого основания при взаимодействии с роданидом аммония образует соль – роданид дикаина – белый кристаллический осадок с определенной Т°пл (130-132оС)
дикаин ∙ НС1 + NH4SCN → дикаин ∙ HSCN↓ + NH4CIТ°пл
3.ГФХреакция на хлорид-ионы
для доказательства наличия сложноэфирной группы можно провести щелочной гидролиз дикаина:

осадок
При подкислении НС1 выделяется осадок п-бутиламинобензойной кислоты, который растворяется в избытке хлороводородной кислоты, а при последующем действии нитрита натрия выпадает осадок N-нитрозопроизводного этой кислоты:

Чистота
ГФХОбщая степень очистки – прозрачность и цветность спиртового раствора, хлориды, кислотность, сульфатная зола, потеря в весе при высушивании.
допустимые: сульфаты, органические примеси в пределах эталона.
по ФС
1посторонние примеси(п-аминобензойную кислоту) методом ТСХ на пластинах "Силуфрол 254", детектируют в УФ свете
2микробиологическая чистота.
Количественное определение
1ФС и ГФХ– нитритометрия, но т.к. дикаин является вторичным ароматическим амином – продукт его взаимодействия с нитритом натрия в среде хлороводородной кислоты N-нитрозопроизводное. Индикатор внутренний – нейтральный красный или тропеолин 00.

2По связанной с азотистым основанием хлороводородной кислоте
А) Алкалиметрическое титрование
Дикаин ∙ НС1 + NaOH → Дикаин ↓ + NaCl + H2O
основание
Индикатор – фенолфталеин, наблюдают за окраской водного слоя.
Б) Аргентометрическое титрование (вариант Фаянса).
Индикатор – бромфеноловый синий, после добавления индикатора
раствор подкисляют разведенной уксусной кислотой до желто-зеленого окрашивания раствора.
Титруют от желто-зеленого до фиолетового окрашивания:
Дикаин ∙ НС1 + AgNO3 → AgCl↓ + Дикаин ∙ HNO3
3Прямое броматометрическое титрование. Индикатор – метиловый красный, разрушаясь избыточной каплей титранта обесцвечивается в точке эквивалентности.
Условия храненияСписок А. В хорошо укупоренной таре. Применение Дикаин – сильное местноанестизирующее средство.
 (zip - application/zip)
(zip - application/zip)